Article Text

Abstract
The incidence of myeloid neoplasms following treatment with poly (ADP-ribose) polymerase inhibitors (PARPi) in patients with ovarian cancer has been gradually increasing over the last few years. The cumulative exposure to PARPi and the improved overall survival of patients with ovarian cancer may represent key underlying explanations behind such trend. Fortunately, the earlier introduction of PARPi in the frontline setting reduces the risk of developing secondary myeloid neoplasms. The etiopathogenesis is still unclear but is likely to be multifactorial. The first 2 years of PARPi exposure seem to be the critical window for the onset of myeloid neoplasms post PARPi, with persistent cytopenia recognized as an early warning sign. Despite intensive treatment strategies, the outcome remains poor. There is an unmet clinical need to learn how to minimize risk, make an early diagnosis, and manage myeloid neoplasms post PARPi. First, decision making regarding the optimal maintenance treatment should avoid a ‘PARPi-for-all’ strategy. PARPi should be used cautiously in cases of high baseline risk for myeloid neoplasms and/or patients who are less likely to have a benefit. Active surveillance, accurate differential diagnosis, and prompt hematological referral are key management pillars. This review discusses what is known on this emerging issue as well as unresolved questions.
- Ovarian Cancer
- Medical Oncology
Statistics from Altmetric.com
Background
With the progressive use of poly (ADP-ribose) polymerase (PARP) inhibitors (PARPi) as maintenance treatment for patients with ovarian cancer, secondary myeloid neoplasms, which were traditionally considered rare delayed adverse events, are gradually emerging as challenging life-threatening toxicities to the point that the Food and Drug Administration (FDA) has raised a warning label. According to the recent 5th edition of the World Health Organization (WHO) classification of hematolymphoid tumors, myeloid neoplasms arising secondary to the exposure to cytotoxic therapies are now known as ‘myeloid neoplasms post cytotoxic therapy’.1 The more precise designation ‘post cytotoxic therapy’, which replaces the older term ‘therapy-related’, reflects a better understanding of the etiopathogenesis and includes the association with new cytotoxic drug classes such as PARPi. Myeloid neoplasms post cytotoxic therapy account for 10–20% of all myeloid neoplasms and include myelodysplastic neoplasms, myelodysplastic/myeloproliferative neoplasms, and acute myeloid leukemia. Myelodysplastic neoplasms are defined as clonal hematopoietic stem cell disorders characterized by unproductive hematopoiesis resulting in peripheral blood cytopenias. These disorders predominantly affect elderly people and might progress to acute myeloid leukemia in up to one-third of cases.2 Their clinical course is typically progressive and relatively resistant to conventional therapies used for de novo myeloid neoplasms. The median life expectancy is around 8 months and the 5-year overall survival is less than 10%.3
The incidence of myeloid neoplasms post PARPi in patients with ovarian cancer has been gradually increasing over the last few years.4 This emerging phenomenon can be explained by several reasons: (1) global population aging; (2) improved overall survival for ovarian cancer patients receiving PARPi, which as a consequence increases the overall treatment burden and widens the time window within which such uncommon events can be detected; (3) longer PARPi exposure; (4) expanding indications for PARPi beyond ovarian cancer; and (5) raised awareness of post-marketing drug safety surveillance. Therefore, there is an urgent clinical need to quickly learn how to reduce the risk, make an early diagnosis, and manage myeloid neoplasms post PARPi. Since transient myelosuppression is among the most frequent adverse events associated with PARPi exposure, physicians must be prepared to properly distinguish it from the more serious secondary hematological malignancies and identify patients most likely to be referred for hematological consultation.
The issue of myeloid neoplasms post PARPi will probably become even more prominent with the new treatment armamentarium on the horizon for ovarian cancer, including PARPi rechallenge and PARPi combination regimens (especially with cell-cycle inhibitors). Moreover, due to the expanding indications for PARPi beyond ovarian cancer, including breast cancer, prostate cancer, and pancreatic cancer, this clinical challenge will soon concern not only gynecologic oncologists but all medical oncologists. This article aims to raise awareness of myeloid neoplasms post PARPi and critically discusses the state-of-the-art knowledge as well as unresolved open issues.
Epidemiology
Data from Clinical Trials
Preliminary data on myeloid neoplasms post PARPi in patients with ovarian cancer enrolled in randomized clinical trials indicated an overall incidence of 0.2–2% (Table 1).5–11 However, this rate became higher after the long-term follow-up analyses from two trials in the recurrent setting. The incidence of niraparib-related myeloid neoplasms nearly doubled after a 5.5-year follow-up (3.5% vs 1.7%) in the NOVA trial (niraparib as maintenance therapy in platinum-sensitive recurrent ovarian cancer regardless of BRCA status),12 and the final analysis of the SOLO2 trial (olaparib as maintenance therapy in BRCA-mutated platinum-sensitive recurrent ovarian cancer) showed an impressive four-fold increase in olaparib-related myeloid neoplasms (8% vs 2%) after approximately 6 years of follow-up.13 On the other hand, the 7-year follow-up safety data from the SOLO1 trial (olaparib as first-line maintenance therapy in BRCA-mutated patients) showed no new cases of myeloid neoplasms with an overall rate that settled at around 1%.14 15 Altogether, these data seem to suggest that the use of PARPi in the recurrent setting is associated with a higher risk of developing late secondary hematological malignancies. This might be explained by the higher amount of genotoxic anti-cancer treatments the patients received compared with the first-line treatment and also by the longer duration of PARPi maintenance in the recurrent setting, where the PARPi is used until progression or unacceptable toxicity compared with up to 2–3 years in first-line treatment. The impact of previous therapies was investigated in a post-hoc analysis of the SOLO2 trial presented at the 2018 annual ASCO meeting, which showed that the incidence of myeloid neoplasms after olaparib increased with the increasing number of prior platinum lines from 0.9% in patients who received two lines to 8% in those who received four or more.16 Recently, a safety meta-analysis of 18 placebo-controlled randomized clinical trials studied the incidence of and mortality caused by myeloid neoplasms in 7307 patients with solid cancers treated with PARPi (4533 PARPi vs 2774 placebo).17 More than 90% of patients were female and the main indication for PARPi was ovarian cancer (85%). The most used PARPi was olaparib (75%) as it was the first PARPi on the market. The incidence of myeloid neoplasms was 0.73% in patients who received PARPi compared with 0.47% in the placebo group. The overall odds ratio (OR) was 2.63 (95% CI 1.13 to 6.14) (p=0.026). A sub-group analysis showed an OR of 1.93 (95% CI 0.68 to 5.49) in the first-line setting and 4.79 (95% CI 1.11 to 20.63) in the recurrent setting, suggesting that both PARPi and prior therapies contribute to the development of myeloid neoplasms.
Incidence of myeloid neoplasms post PARPi in patients with ovarian cancer enrolled in pivotal randomized clinical trials
Nevertheless, data from clinical trials present several limitations. Due to the rigorous inclusion/exclusion criteria, the study population is often not representative of the entire general population which, for instance, also includes individuals with a high burden of co-morbidities. Moreover, the sample size is relatively small and the duration of follow-up is often too short to accurately assess rare events.
Real-World Data
Given the non-negligible limitations associated with clinical trials, huge efforts have recently been made to provide real-world evidence. Interesting data have been published in the last 3 years both from pharmacovigilance databases and from research institutions.
Pharmacovigilance Studies
Since 2020, several pharmacovigilance studies have been published using international databases such as the Food and Drug Administration Adverse Event Reporting System (FAERS), the WHO database VigiBase, and the European Medicines Agency database EudraVigilance (EV).4 17–20 Ma et al first published a pharmacovigilance analysis of the FAERS database including 319 patients and showed that the number of myeloid neoplasms post PARPi increased exponentially from 2015 to 2019.4 Over 90% of patients were female and ovarian cancer was the main indication in about three-quarters of the cases. The median time to onset was around 15 months. Olaparib was the most used PARPi (80%) and its OR was 48.03 compared with 6.58 and 2.23 for niraparib and rucaparib, respectively. This study reported death in 32% of cases but mortality outcomes were better defined in a subsequent analysis of the FAERS database by Matsuo et al,18 which addressed 403 hematological malignancies and showed that 28.3% of myelodysplastic syndromes and 40.8% of acute myeloid leukemias were fatal. Similar data were derived from the study of the VigiBase database by Morice et al,17 which included 178 cases of myeloid neoplasms and reported a median PARPi treatment duration, median event onset, and mortality rate of 9.8 months, 17.8 months, and 45%, respectively. Interestingly, this analysis showed that 40% of patients experienced a prior cytopenia, with anemia being the most frequent type (34%), at a median of 7 months from PARPi initiation before developing myeloid neoplasms. Therefore, particular attention should be paid to cytopenias occurring several months after PARPi initiation, as they might represent an alarm bell for the diagnosis of myeloid neoplasms. In June 2022, Zhao et al published an updated analysis of myeloid neoplasms post PARPi reported between 2014 and 2021 from both the FAERS and EV databases, which again confirmed previous findings.19 The median time to onset was around 7 months for myelodysplastic syndromes and 12 months for acute myeloid leukemia. The overall mortality rates of myelodysplastic syndromes and acute myeloid leukemia were 39% and 45% in the FAERS database and 32% and 34% in the EV database, respectively. Again, the OR for olaparib (40.49) was significantly higher compared with the other PARPi niraparib and rucaparib (5.29 and 4.22, respectively).
All these recent pharmacovigilance studies suggest a higher rate of myeloid neoplasms post PARPi in the real-world setting compared with randomized clinical trials. More than 80% of PARPi were used for patients with ovarian cancer and, among all PARPi, olaparib appeared to have the strongest association with myeloid neoplasms. Overall, the outcomes were extremely poor with a reported mortality rate ranging from 30% to 45%. The first 2 years after starting PARPi were the ‘critical pharmacovigilance window’, where the vast majority of hematological malignancies occurred. However, this median time to onset of myeloid neoplasms may be biased since patients with ovarian cancer frequently relapse within 2 years.
Real-world data retrieved from pharmacovigilance databases are not without limitations. The exact incidence of myeloid neoplasms post PARPi cannot be calculated lacking the total number of patients under PARPi and data are tainted with relevant reporting biases. Indeed, the reporting process is arbitrary, and this could have led to an underestimation of the real incidence. The sources are heterogeneous, including both healthcare and non-healthcare practitioners, and the accuracy of the content was not rigorously verified. Finally, important data were missing, including PARPi dose, maintenance duration, PARPi line, BRCA/HRD status, number and type of prior therapies, onset timing of myeloid neoplasms, and cause of death.
Data from Research Institutions
The first real-life data on myeloid neoplasms post PARPi in patients with ovarian cancer outside pharmacovigilance studies were published in 2020 and then updated in 2021 by the Institute Gustave Roussy.21 They evaluated 20 patients with ovarian cancer with myeloid neoplasms post PARPi and reported BRCA mutations in 75% of cases, mutations in DNA damage repair (DDR) genes (TP53 or PPM1D) in 83%, unfavorable cytogenetic features in 100% (of which 95% were complex karyotype), and a poor prognosis (median overall survival of 4.3 months). The median PARPi exposure and time to onset of myeloid neoplasms were 17 months and 2 years, respectively. The European Institute of Oncology studied 182 patients with ovarian cancer treated with various PARPi, of whom 16 (8.7%) developed secondary hematological malignancies (12 myelodysplastic syndrome and 4 acute myeloid leukemia).22 23 This incidence of 8.7% was the first from a real-life setting and is in line with the 8% rate retrieved from the final analysis of the SOLO2 trial. All patients experienced persistent cytopenia after PARPi suspension. The most common cytogenetic abnormalities were structural rearrangements of chromosomes 5, 7, and 17 and/or complex karyotypes. TP53 gene was mutated in over 90% of patients. Despite intensive treatment regimens, the outcomes were extremely poor with a mortality rate of 75% after a median follow-up of 6.8 months. In 2022, two other series on this topic were published. The Mayo Clinic identified nine cases of myeloid neoplasms in a cohort of 583 PARPi-treated patients (1.5%) and their clinical, cytogenetic, and molecular findings are in agreement with previous studies.24 The median time to onset of myeloid neoplasms was 19 months and the mortality rate was 44%. Once again, TP53 was the most commonly involved gene. Finally, another recently published series included 13 patients with ovarian cancer with myeloid neoplasms (4.3%) in a cohort of 300 patients exposed to PARPi.25 All patients experienced a prior persistent cytopenia, showed an unfavorable karyotype, and shared a common molecular profile. They all reported BRCA and TP53 mutations and, interestingly, 69% of them also harbored TET2 and/or DNMT3A gene mutations, which have been shown to display a pre-leukemic potential. The median time to onset was 12 months and the mortality rate after a median follow-up of 5 months was 69%.
The pooled data from these research institutions reported an incidence rate of 1.5–8.7%, a median time of PARPi exposure of 10–17 months, a median time to onset of myeloid neoplasms (from PARPi initiation) of 7–24 months, and an overall mortality rate ranging from 30% to 75%. Furthermore, these four studies shared some common findings that constitute typical features of myeloid neoplasms post PARPi: (1) complex karyotype with frequent involvement of chromosomes 5, 7, and 17; (2) predominant TP53 and other DDR mutations, suggesting their central role in the pathogenesis; (3) relatively rapid onset, in contrast to other agents such as alkylating agents, which usually lead to myeloid malignancies within 5–10 years. These characteristics seem to be directly linked to the specific mechanism of action of PARPi and thus differ from de novo or other treatment-related myeloid neoplasms.
Pathogenesis
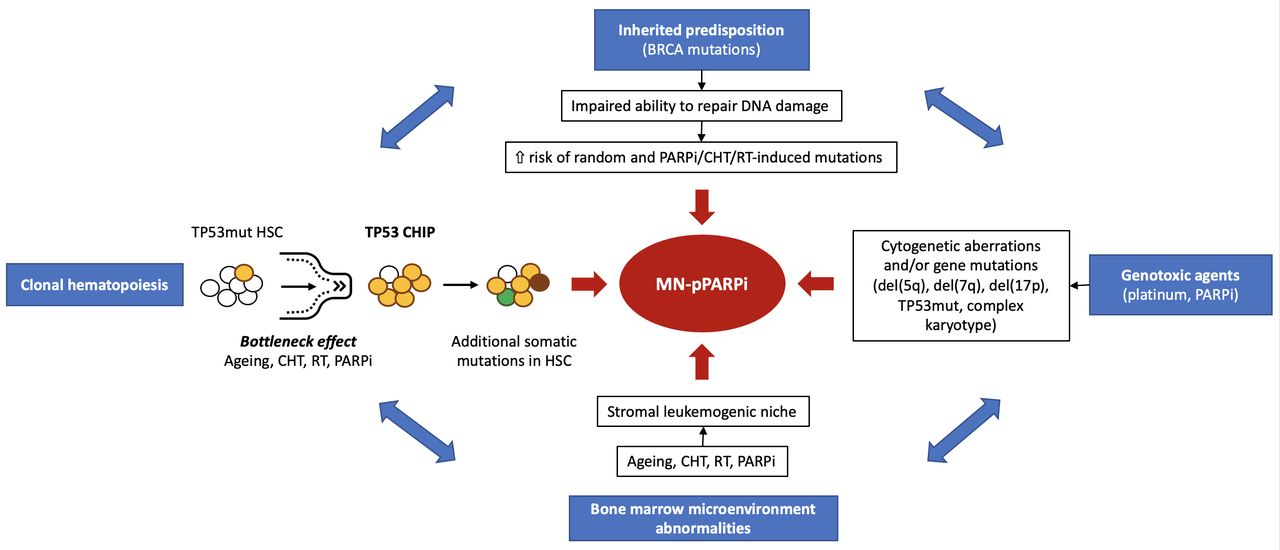
The etiopathogenesis of myeloid neoplasms post PARPi is not completely understood and needs further investigation. However, four interrelated key pathways seem to contribute to their development: (1) inherited predisposition; (2) direct DNA damage induced by cytotoxic agents; (3) clonal hematopoiesis; and (4) impaired bone marrow environment (Figure 1).26 27

The pathogenesis of myeloid neoplasms post PARPi showing four main interrelated drivers. CHIP, clonal hematopoiesis of indeterminate potential; CHT, chemotherapy; HSC, hematopoietic stem cell; MN-pPARPi, myeloid neoplasms post PARPi; PARPi, poly (ADP-ribose) polymerase inhibitor; RT, radiotherapy; TP53mut, TP53 gene mutations.
Inherited Predisposition
Women with germline genetic mutations that impair the correct functioning of genes involved in DDR mechanisms, such as BRCA (~15% of patients with ovarian cancer) or homologous recombination (HR) pathway, have a higher a priori risk of acquiring subsequent sporadic mutations, eventually leading to multiple independent secondary malignancies.28 Moreover, due to the reduced ability to repair DNA damage, these patients show a higher susceptibility to chemotherapy and radiotherapy-induced alterations and thus again to secondary malignancies. Older age and higher co-morbidity burden also play a pivotal role as pre-existing factors. A post hoc analysis of the SOLO2 trial showed a higher frequency of myeloid neoplasms in BRCA-mutated patients with recurrent platinum-sensitive ovarian cancer.29
DNA Damage Induced by Mutagenic Agents
The exposure to genotoxic agents, including platinum salts and PARPi, is itself associated with an increased risk of accumulating DNA damage and developing treatment-related myeloid neoplasms. Indeed, the effects of DNA-damaging agents used as anti-cancer treatments extend also to healthy rapidly dividing cells such as hematopoietic cells in the bone marrow.30 The cytogenetic abnormalities and/or gene mutations most commonly observed so far in myeloid neoplasms post PARPi include chromosome 5q deletion, partial or complete loss of chromosome 7, rearrangements of chromosome 17, TP53 mutations, and/or complex karyotype.
Using data from large population-based databases in the USA including 23 862 patients with ovarian cancer, Shenolikar et al reported an incidence rate of secondary myeloid neoplasms following antineoplastic treatments for ovarian cancer of 2.77 per 1000 person-years.31 This incidence was higher in patients exposed to DNA-damaging therapies. Platinum, alkylating agents, and topoisomerase inhibitors showed the strongest association with the onset of myeloid neoplasms and the duration of treatment exposure was a significant risk factor. These data suggest that there is a baseline risk of developing treatment-related myeloid neoplasms due to exposure to DNA-damaging therapies even before the use of later line therapies such as PARPi. In 2019, Morton et al published a population-based cohort study using cancer registries from the Surveillance, Epidemiology, and End Results (SEER) database, showing the association of chemotherapy for solid tumors with the development of therapy-related myeloid neoplasms.32 The analysis included 700 612 patients with a mean age of 64.3 years and 1619 cases of myeloid neoplasms. The relative risk (RR) of developing therapy-related myeloid neoplasms ranged from 1.5 to ≥10 across various solid tumors and, in particular, it was 5.8 for ovarian cancer.
Clonal Hematopoiesis
PARPi may act as a driver for clonal hematopoiesis of indeterminate potential. This is defined as the clonal selection of a blood sub-population from a single hematopoietic stem cell or progenitor cell with acquired somatic mutations that gave a competitive advantage in hematopoiesis over other stem/progenitor cells. These clonally shared somatic abnormalities involve genes potentially responsible for myeloid malignancies and can be detected at a variant allele frequency of at least 1–2% in the peripheral blood cells of healthy individuals.33 Clonal hematopoiesis of indeterminate potential likely represents a pre-cancerous clonal expansion with a huge potential for leukemic transformation (13-fold increased risk). Its prevalence physiologically increases with aging, correlates with exposure to mutagenic agents (chemotherapy, radiotherapy, smoking), and is 5–10% higher in patients with solid tumors compared with the general population. Clonal hematopoiesis of indeterminate potential variants in DDR genes, including TP53, PPM1D, and CHEK2, specifically impart survival advantage and preferentially expand during platinum-based chemotherapy and PARPi.34 PARP enzymes and p53 play a crucial role in DNA repair and genomic stability, and TP53-mutated cells highly depend on PARP activity for repairing DNA double-strand breaks. Therefore, PARP inhibition facilitates the selection of hematopoietic stem cells with pre-existing TP53 clonal hematopoiesis of indeterminate potential variants (expanded after platinum) through a ‘bottleneck’ effect, leading to subsequent genetic instability and accumulation of additional somatic mutations, potentially causing leukemogenic transformation. Ultimately, PARPi, prolonged exposure to prior cytotoxic agents, and the presence of TP53-mutated clones synergistically interact and contribute to the development of myeloid neoplasms post PARPi, even more in the context of inherited predisposition (BRCA mutations).
Bolton et al first suggested that PARPi, along with a multifactorial background (prior therapies and germline BRCA mutation), promote the expansion of DDR clonal hematopoiesis of indeterminate potential variants.35 In a prospective analysis of cancer patients, the growth rate of DDR clonal hematopoiesis of indeterminate potential variants was significantly higher among those treated with PARPi (median +2.8% increase in variant allele frequency per year) compared with other cytotoxic therapies (+1% per year, p=0.04) or untreated patients (+0.08% per year, p=0.02). Intriguingly, a recent retrospective genetic study evaluated the risk of myeloid neoplasms associated with TP53 clonal hematopoiesis of indeterminate potential variants in 1052 patients with ovarian cancer treated with rucaparib from the ARIEL2 and ARIEL3 trials (rucaparib as maintenance therapy in platinum-sensitive recurrent ovarian cancer, regardless of BRCA status).36 The incidence of myeloid neoplasms was 2.1%. Peripheral blood cell samples collected before rucaparib initiation from 20 cases of myelodysplastic syndrome were compared with those from 44 controls. This study showed that pre-PARPi TP53 clonal hematopoiesis of indeterminate potential variants was a risk factor for developing secondary myeloid neoplasms after PARPi exposure. Moreover, the amount of TP53 clonal hematopoiesis of indeterminate potential variants was higher in patients with longer overall exposure to prior platinum, thus suggesting a background for the higher rate of myeloid neoplasms after multiple platinum lines in the SOLO2 post hoc analysis. Martin et al reported a higher rate of clonal hematopoiesis of indeterminate potential variants among patients with ovarian cancer free of myeloid neoplasms who received PARPi compared with the non-PARPi group (78% vs 39%; p=0.018).21 Moreover, 67% of patients treated with PARPi harbored a mutation in DDR genes compared with 17% in those without PARPi (p=0.002). Overall, these data support the hypothesis that PARPi may serve as a selective pressure on the clonal expansion of DDR mutations, which are known to determine a poor prognosis.
Impaired Bone Marrow Environment
Alterations in the bone marrow microenvironment represent another key mediator of the pathogenesis of myeloid neoplasms. Mesenchymal stromal cells and endothelial cells critically contribute to the overall stability (quiescence, proliferation, and migration) of stem cell pools and the regulation of physiologic hematopoiesis through the production of signaling molecules (growth factors, cytokines, chemokines) and receptors. PARPi—along with chemotherapy, radiotherapy, and aging, among other leukemogenic risk factors—cause significant damage to the bone marrow stromal milieu and epigenetic modifications which further accelerate DNA damage and clonal hematopoiesis, hence favoring the initiation and progression of secondary hematological malignancies.
Strategic Management: a Four-Step Approach
Gynecologic oncologists and medical oncologists should follow a four-step process to reduce risk, make an early diagnosis, and properly manage myeloid neoplasms post PARPi: (1) tailored selection of PARPi users; (2) proactive surveillance and monitoring; (3) differential diagnosis with transient PARPi-related myelosuppression; and (4) prompt referral to a hematologist.
Is PARPi Maintenance the Right Choice?
The advent of PARPi certainly marked a promising new era in the treatment of ovarian cancer. However, this enthusiasm should not be used to mislead into adopting a single ‘PARPi-for-all’ strategy. Decision making regarding the best maintenance option could be demanding in daily practice, especially in the HR-proficient setting, and multiple factors apart from the mutational status need to be considered. Answering this question goes beyond the scope of this paper but represents the first step to prioritize PARPi use where the risk–benefit ratio is favorable.
What is the Appropriate Surveillance Strategy under PARPi?
Active surveillance and monitoring during PARPi exposure are essential to catch the warning signals as soon as possible, without underestimating a serious complication because it is erroneously regarded as extremely rare. Appropriate counseling on these life-threatening toxicities is imperative before starting PARPi, and patients should be educated on how to recognize suspicious symptoms early. Screening for pre-existing risk factors (prior cytotoxic therapies, older age, germline BRCA mutations, blood disorders, family history, and smoking) is pivotal as it helps to identify patients at higher risk of developing secondary malignancies, therefore requiring closer surveillance. The follow-up program includes a baseline complete blood count before PARPi initiation with subsequent regular monthly monitoring for the first year (and then at the physician’s discretion). Notably, the surveillance should not be stopped immediately after PARPi discontinuation but instead continued for at least 7 months based on the median time to onset of myeloid neoplasms reported in both clinical trials and real-world pharmacovigilance studies.
How Can we Distinguish Myeloid Neoplasms Post PARPi from Transient Hematotoxicities?
Hematological adverse events are among the most frequent toxicities associated with PARPi and the predominant cause of dose reduction and dose interruption.37 These complications result from transient PARPi-caused myelosuppression and typically occur within the first 3 months with a mild-to-moderate severity grade. Their management is outlined in Figure 2. Briefly, grade 1 anemia or neutropenia can be strictly monitored with weekly blood cell counts while patients continue PARPi treatment. In case of grade 2–4 anemia/neutropenia and any-grade thrombocytopenia, PARPi should be temporarily withheld until blood parameters resolve to grade 0–1 and up to a maximum of 28 days. PARPi can be resumed at the same dose (first adverse event) or a reduced dose in case of recurrent adverse events or niraparib as PARPi type. PARPi must be discontinued if hematological toxicity persists after 4 weeks from PARPi suspension or if the PARPi was already at the lowest dose.38 On the other hand, myeloid neoplasms post PARPi are rare but severe and typically occur as delayed toxicity 7–24 months after PARPi initiation, although this is not always the case as they can occasionally present soon after starting PARPi. Notably, PARPi suspension acts as a sort of ‘litmus test’, allowing myeloid neoplasms to be distinguished from non-malignant blood disorders. Indeed, while non-malignant disorders fully resolve after PARPi suspension, the symptoms of myeloid neoplasms persist after 4 weeks (Table 2). Furthermore, the differential diagnosis should also rule out other causes of cytopenias including alcohol or other drugs, sideropenic anemia, hemolytic anemia, vitamin (B12 or folate) deficiency, autoimmune disorders, renal failure, and chronic infections.

{kind=link}
{kind=link}
Outline of the management of PARPi-related hematological adverse events. CBC, cell blood count; Hb, hemoglobin; neu, neutrophils; PARPi, poly (ADP-ribose) polymerase inhibitor; PLTs, platelets.
Differential diagnosis between non-malignant hematological adverse events associated with PARPi and myeloid neoplasms post PARPi
When should Patients be Referred to a Hematologist?
Physicians should promptly refer patients to a hematologist for careful diagnostic procedures whenever one of the following scenarios occurs: (1) before starting PARPi in case of pre-existing risk factors; (2) unexplained acute and/or severe cytopenia (even soon after PARPi initiation); (3) blood parameters remain clinically abnormal 4 weeks after dose interruption; (4) recurrent cytopenia despite dose modifications; (5) single or multi-lineage cytopenias after the first 3 months of PARPi and especially after 7 months from PARPi exposure. Ultimately, every unusual cytopenia, even if minor, should not be minimized as it might represent an early warning sign for the onset of myeloid neoplasms.
Diagnosis and treatment
The detailed discussion of the diagnosis and treatment of myeloid neoplasms goes beyond the purposes of the present review and can be found in dedicated international guidelines.2 39 Here we provide only a brief discussion specifically focused on myeloid neoplasms post PARPi. The diagnosis is based, as for all myeloid neoplasms, on peripheral blood and bone marrow analyses: (1) blood cell count showing cytopenias (mostly anemia) and other laboratory parameters (ferritin, transferrin, reticulocyte count, B9/B12 vitamins, creatinine, haptoglobin), which are useful for the differential diagnosis with other non-malignant diseases; (2) cytomorphology (hypercellular or sometimes hypocellular bone marrow with dysplastic features in ≥10% of cells, with or without an excess of blasts); (3) cytogenetics (del(5q)/−5, del(7q)/−7, rearrangements of chromosome 17, and/or complex karyotype); (4) molecular genetics (BRCA, TP53); (5) detection of clonal hematopoiesis of indeterminate potential variants; and (6) flow cytometry.
The prognosis for myeloid neoplasms depends on the marrow blast count, the number and severity of cytopenias and dysplasias, and cytogenetic aberrations. Myeloid neoplasms post PARPi are characterized by high genetic complexity compared with de novo myeloid neoplasms and this could explain the extremely poor outcomes despite the intensive therapies. Management is personalized in a risk-adapted manner and based on prognostic risk factors according to the revised International Prognostic Score System (IPSS-R), as well as on patient age, co-morbidities, and frailty score. The treatment strategy varies from symptomatic therapies for cytopenias in lower-risk patients (mainly transfusions, growth factors, and antibiotics) to hypomethylating agents (HMA) such as azacytidine or decitabine, standard chemotherapy (cytarabine with idarubicin or fludarabine), or allogeneic hematopoietic cell transplantation (preceded or not by induction chemotherapy or HMA) in higher-risk patients.
It is important to mention that treatment options for patients with ovarian cancer with secondary myeloid neoplasms are much more limited and also the oncologic treatment for ovarian cancer is not always feasible if needed. If at least one of the two tumors is in remission, specific treatment of the active one can be attempted. However, if both neoplasms are progressing and require treatment, it is difficult to establish which is the priority and there are no evidence-based guidelines to support specialists in handling these puzzling cases. Since ovarian cancer and myeloid neoplasms represent two completely different cancers, finding a therapeutic strategy that fits both cancer types without serious toxicity is challenging. One attempt might be to control ovarian cancer with radiotherapy while treating myeloid malignancy, but for the moment this is only anectodal.23 The optimal management remains an unmet clinical need and should be discussed in referral centers with expertise and experience in this field. Multidisciplinary collaboration and patient-centered decision making are recommended.
Open Questions and Future Perspectives
Several issues remain unresolved and warrant further research in the future. First, there is an unmet need for more real-life data with longer follow-up periods to properly assess myeloid neoplasms. Predictive biomarkers of the putative risk of developing myeloid neoplasms after PARPi initiation are urgently required as they might help to properly stratify patients based on their risk and tailor their surveillance program during PARPi exposure. A better understanding of the role of clonal hematopoiesis is essential. The screening for TP53 (or other DDR) clonal hematopoiesis of indeterminate potential variants from peripheral blood cell samples collected before starting PARPi treatment might represent a promising research field and offer opportunities for prevention. Prospective screening for clonal hematopoiesis-associated mutations is part of the protocol of ongoing trials with PARPi, such as the AGO-OVAR 28 trial.40 The contribution of BRCA/HRD status remains to be clarified. Indeed, germline BRCA-mutated patients are predisposed to multiple platinum-sensitive cancers and are more likely to have received multiple lines with platinum and alkylating agents; therefore, it is difficult to distinguish the impact of prior chemotherapy. More comprehensive genetic analysis beyond BRCA, including TET2, DNMT3A, and FLT3, might be useful in identifying patients at higher risk of developing myeloid neoplasms.
Notably, PARPi do not seem to be the only ones responsible for secondary hematological malignancies in patients with ovarian cancer, but it is more likely that other risk factors play a pivotal role in this scenario. The comparison of long-term safety data from the SOLO2 and SOLO1 trials underlined the remarkable impact of prior platinum lines. A recent meta-analysis suggested that heavily pre-treated patients as well as those harboring a BRCA mutation have a higher a priori risk of developing secondary malignancies, regardless of PARPi treatment, and it is unclear what PARPi add to this baseline risk.41 It is also possible that the higher incidence of myeloid neoplasms seen in patients with ovarian cancer treated with PARPi is actually due to the increased overall survival widening the time window for the development of secondary malignancies after exposure to multiple treatments. Currently, it is not possible to establish which factor contributes most to the development of myeloid neoplasms post PARPi—whether PARPi, prior platinum-based chemotherapies, or the synergistic interaction between multiple drivers. The recent introduction of PARPi as first-line maintenance for patients with ovarian cancer will probably clarify the real role of PARPi in leukemogenesis, unbiased by prior cumulative chemotherapy. Moreover, it remains unclear to what extent the duration of PARPi exposure (as well as other DNA-damaging agents) is decisive in the pathogenesis of myeloid neoplasms.
Since PARPi are not all equal but display different pharmacokinetic and pharmacodynamic profiles, discrepancies between various PARPi should be investigated to identify the leukemogenic potential of each PARPi (drug-specific risk).42 Olaparib has shown the strongest association with secondary myeloid neoplasms among all PARPi. However, although the rationale behind this could rely on its peculiar pharmacodynamics,30 43 44 potential biases should be considered, including the fact that olaparib was the first PARPi on the market and has more treatment indications compared with other PARPi, and thus we have more evidence available and longer follow-up times while data for other PARPi might be immature. In addition, since mutations in BRCA and HR repair genes have been associated with an increased risk of myeloid neoplasms and olaparib is specifically used for BRCA/HR-deficient patients, the contribution of these mutations to the higher risk of myeloid neoplasms reported for olaparib should be clarified.
The risk of developing myeloid neoplasms associated with emerging PARPi combined regimens (with anti-angiogenics, immune checkpoint inhibitors, cell-cycle inhibitors) and PARPi rechallenge is mostly unknown and needs prompt consideration. In the final analysis (5-year follow-up) of the PAOLA1 trial (olaparib plus bevacizumab as first-line maintenance), the incidence of myeloid neoplasms remained low and balanced between arms.45 However, safety data from other combinations and from the OReO trial (olaparib rechallenge in platinum-sensitive recurrent ovarian cancer, regardless of BRCA status) are awaited. PARPi combination regimens will probably become the new key strategy, especially in the recurrent setting. Indeed, the detrimental overall survival data from the ARIEL4 (rucaparib as monotherapy in somatic and/or germline BRCA-mutated patients)46 and SOLO3 (olaparib as monotherapy in germline BRCA-mutated patients)47 trials have led to the withdrawal of the PARPi indication (as single agents) to treat late-line ovarian cancer. Moreover, while survival results from ARIEL348 support the use of PARPi as second-line maintenance treatment for platinum-sensitive recurrent ovarian cancer regardless of BRCA status, the updated overall survival analysis from the NOVA trial12 warrants caution in using niraparib for the sub-set of BRCA wild-type patients.
Finally, increasing attention is being paid to next generation, highly selective PARPi. The PARP family consists of 17 enzymes that differ greatly in structure and functions. The PARPi currently used in clinical practice lack selectivity and target PARP enzymes indiscriminately and often redundantly. Pre-clinical data suggest that the carcinogenic role of each PARP enzyme might be cancer-specific, so a more selective PARPi could be used to treat each cancer type, thus increasing the effectiveness while minimizing the adverse events. In this regard, the PARP1-specific inhibitor AZD5305 has been tested in in vitro and in vivo models and was found to retain the therapeutic efficacy of non-selective PARPi while reducing hematotoxicity.49 AZD5305 is currently being investigated in the phase I PETRA trial (NCT04644068), both as monotherapy and in combination with anti-cancer agents in patients with advanced solid malignancies.
Summary statements
The survival benefit of PARPi maintenance for patients with ovarian cancer is out of the question, at least in the BRCA-mutated/HR-deficient setting.
Myeloid neoplasms post PARPi in patients with ovarian cancer are gradually emerging as life-threatening late toxicities and should not be minimized.
The first 2 years of PARPi exposure are the critical window for the onset of myeloid neoplasms and persistent cytopenia has been recognized as an early warning sign.
Active surveillance, differential diagnosis, and prompt hematological referral are crucial.
PARPi should be used in the first-line setting, not only because of their efficacy but also to reduce the risk of myeloid neoplasms leading to an improved risk–benefit ratio.
Decision making regarding the optimal maintenance treatment should avoid a ‘PARPi-for-all’ strategy.
PARPi should be used cautiously in patients with a baseline risk for myeloid neoplasms and/or those who are less likely to have a significant benefit, as in the first-line HR-proficient setting.
Further research with long-term real-world safety data is needed to shed more light on this fatal toxicity, define its actual incidence, and find risk-reducing measures to preserve patients at higher risk.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
References
Footnotes
Correction notice This article has been corrected since it was first published. The author affiliations have been updated.
Contributors GC: Conceptualization, data curation, investigation, methodology, resources, software, validation, visualization, writing - original draft, writing - review and editing. FG, GP, ML, SD, IP: data curation, methodology, validation, visualization, writing - review and editing. NC: project administration, validation, supervision; writing - review and editing. All authors have read and agreed to the published version of the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests NC reports fees for advisory board membership for AstraZeneca, Clovis Oncology, Eisai, GSK, Immunogen, Mersana, MSD/Merck, Nuvation Bio, Onxerna, Pfizer, Pieris, Roche; fees as an invited speaker for AstraZeneca, Novartis, Clovis Oncology, GSK, MSD/Merck; institutional research grants from AstraZeneca, and Roche. She has also reported non-remunerated activities as a member of the ESMO Guidelines Steering Committee and chair of the Scientific Committee of ACTO (Alleanza Contro il Tumore Ovarico). All other authors declare no conflicts of interest.
Provenance and peer review Not commissioned; externally peer reviewed.