
Hsp90: A New Player in DNA Repair?




1
Department of Sciences, Roma Tre University, Viale Guglielmo Marconi 446, Roma I-00146, Italy
2
Istituto Nazionale di Biostrutture e Biosistemi, Viale Medaglie d’Oro 305, Roma I-00136, Italy
*
Author to whom correspondence should be addressed.
Biomolecules 2015, 5(4), 2589-2618; https://doi.org/10.3390/biom5042589
Submission received: 14 July 2015
/
Revised: 8 September 2015
/
Accepted: 10 September 2015
/
Published: 16 October 2015
(This article belongs to the Special Issue DNA Damage Response)
Abstract
:Heat shock protein 90 (Hsp90) is an evolutionary conserved molecular chaperone that, together with Hsp70 and co-chaperones makes up the Hsp90 chaperone machinery, stabilizing and activating more than 200 proteins, involved in protein homeostasis (i.e., proteostasis), transcriptional regulation, chromatin remodeling, and DNA repair. Cells respond to DNA damage by activating complex DNA damage response (DDR) pathways that include: (i) cell cycle arrest; (ii) transcriptional and post-translational activation of a subset of genes, including those associated with DNA repair; and (iii) triggering of programmed cell death. The efficacy of the DDR pathways is influenced by the nuclear levels of DNA repair proteins, which are regulated by balancing between protein synthesis and degradation as well as by nuclear import and export. The inability to respond properly to either DNA damage or to DNA repair leads to genetic instability, which in turn may enhance the rate of cancer development. Multiple components of the DNA double strand breaks repair machinery, including BRCA1, BRCA2, CHK1, DNA-PKcs, FANCA, and the MRE11/RAD50/NBN complex, have been described to be client proteins of Hsp90, which acts as a regulator of the diverse DDR pathways. Inhibition of Hsp90 actions leads to the altered localization and stabilization of DDR proteins after DNA damage and may represent a cell-specific and tumor-selective radiosensibilizer. Here, the role of Hsp90-dependent molecular mechanisms involved in cancer onset and in the maintenance of the genome integrity is discussed and highlighted.
1. Introduction
Cellular protein homeostasis, also termed proteostasis, regulates protein folding and functions in order to maintain the well-being condition of both the cells and the organism; indeed, protein misfolding and unfolding are associated to several human disease [1]. Molecular chaperones and protein remodeling factors maintain proteostasis, mitigating the life-threatening effects of endogenous and exogenous stressors on the proteome [2].
Heat shock protein 90 (Hsp90) is an evolutionary conserved molecular chaperone that, together with Hsp70 and co-chaperones, makes up the Hsp90 chaperone machinery, which stabilizes and activates more than 200 proteins in mammalian cells [3,4,5,6,7]. Two major cytoplasmic isoforms of Hsp90 are known, the inducible/major form Hsp90α and the constitutive/minor form Hsp90β [8]. One of the main structural differences is that the α form readily dimerizes, whereas the β form undergoes dimerization with much less efficiency (see paragraph 3). Since the isolation of Hsp90 isoforms is rather difficult, most studies have been carried out with the mixture of α and β Hsp90 isoforms [9].
Hsp90 is one of the most abundant and conserved molecular chaperones playing an essential role in eukaryotic cells proteostasis. Indeed, Hsp90 is involved in cellular homeostasis, transcriptional regulation, chromatin remodeling, and DNA repair. The ability of the Hsp90 chaperone machine to correlate protein assembly to protein degradation represents a quality control mechanism and provides plasticity for dynamic protein complexes [10,11].
Differently from other chaperones, Hsp90 is not required for de novo protein folding, but rather facilitates the final maturation of specific proteins termed clients. Indeed, most Hsp90 clients must be properly folded in order to interact successfully with their binding partners. Therefore, the Hsp90 chaperone machinery plays a key role in orchestrating the spatial and temporal order of protein interactions [2,3,12]. To ensure the proper protein assembly, the Hsp90 chaperone machinery performs three main functions under normal conditions: (i) it specifically interacts with a vast array of clients through adapter co-chaperones (e.g., p23 and Cdc37); (ii) it stabilizes specific folding intermediates that allows clients to interact with specific binding partners; and (iii) it regulates the ubiquitin-mediated proteasome degradation [2,10,11,13,14].
The Hsp90 activity is regulated at several levels, including the ATPase cycle, the association with conformation-specific co-chaperones, and post-translational modifications [15,16]. Co-chaperones modulate client protein recognition by Hsp90, regulate the ATPase activity of Hsp90, and modulate the client biochemical activities. Among the 20 co-chaperons identified, so far the most important are Cdc37, playing a pivotal role in cell cycle regulation, p23, being a major player in the DNA repair processes, and Aha1, activating the Hsp90 ATPase activity [17,18]. Hsp90 is one of the major eukaryotic phosphoprotein, changes in its phosphorylation status having a deep impact on the chaperone functions [19,20].
Under physiological conditions, Hsp90 represents ~1%–2% of the total cellular protein content, being pivotal to buffer proteostasis against environmental stress [21,22]. Under extreme environmental conditions, the chaperone reservoir can be rapidly exhausted with consequent changes in the functions of Hsp90 clients, thus influencing human health, diseases onset, and evolutionary processes [2]. Because Hsp90 is the molecular chaperone of numerous oncoproteins, it is considered a crucial facilitator of oncogene addiction and represents a validated anti-cancer drug target (see paragraph 2) [23,24]. Indeed, inhibition of the ATPase activity of Hsp90 impairs client protein recognition, thus causing clients degradation [10,25,26].
2. Hsp90 and Cancer
Hsp90 is implicated in many pathological conditions, such as ischemia, reperfusion, infections, neurodegenerative diseases, and particularly cancer [27,28,29,30,31,32,33,34]. Indeed, it is well known that many Hsp90 clients are: (i) oncogenic proteins (e.g., EGFR and Her2/HerbB2); (ii) signaling proteins (e.g., Akt/PKB, Raf1, IKK, p53, v-Src, and HIF-1α); (iii) cell cycle regulators (e.g., Cdk4/6); (iv) chimeric signaling proteins (e.g., Bcr-Abl); and (v) hormone receptors (e.g., estrogen and glucocorticoid receptors) [35,36,37,38,39,40,41].
The essential functions of chaperones are subverted during oncogenesis, contributing to malignant transformation and to the rapid somatic evolution. The increased expression of one or more chaperones above the level observed in normal tissues is a common feature of both solid tumors and hematological malignancies [30,33,42]. The increased abundance of chaperones in advanced cancers reflects an appropriate cytoprotective stress response to the hostile hypoxic, acidotic, and nutrient-deprived microenvironment. At the molecular level, the increased chaperone activities allows tumor cells to cope with the imbalanced signaling that is associated with neoplastic transformation, and thereby escape the apoptotic death that would normally ensue. Indeed, the impairment of apoptotic signaling is a common characteristic of cancer cells, since it facilitates survival and expansion thus rendering cells independent of normal regulatory factors and resistant to host defense mechanisms and both chemotherapy and radiotherapy. Moreover, the ability of cancer cells to repair DNA allows them to survive from the DNA damage induced by chemotherapeutic agents or radiation [30,33,42].
In cancer cells, Hsp90 and its co-chaperones form a super-chaperone complex, i.e., the “activated state” of the protein, and the client proteins display a stable association with Hsp90. By contrast, under normal conditions clients interact with low-affinity with Hsp90 since the super-chaperone complex is not formed, Hsp90 being in the so-called “latent state” [43]. The expression of Hsp90 is 2- to 10-fold higher in tumor cells than normal cells [44]. Therefore Hsp90 has become an attractive therapeutic target in cancer treatment, since its inhibition can induce the degradation of clients and promoting enhanced host natural killer cell-mediated tumor killing [41,45,46,47].
Hsp90 levels significantly increase in medulloblastoma [48] and in breast cancer (particularly in ductal carcinomas) [49]. In colorectal cancer, Hsp90 promotes epithelial to mesenchymal transition, invasion, and migration, both in vitro and in vivo, because of HIF-1α or NF-κB overexpression [50]. Moreover, Hsp90 is an essential regulator of EphA2 stability and signaling [51], which is highly expressed in several cancer cells and is recognized by the host as a self-protein, thus limiting the ability of CD8+ T cells to recognize and kill the tumor. Noteworthy, the 17-DMAG-dependent inhibition of Hsp90 affects EphA2 stability, improving the in vivo anti-tumor activity of Hsp90 [52].
Pre-clinical studies attest the potential of Hsp90 inhibition in inducing tumor growth inhibition, in reducing metastatic potential, and in sensibilizing tumors to the effect of other therapies, possibly as a consequence of the inhibition of DSBs repair and cell cycle checkpoint activation [23,33,35,53,54,55,56,57,58,59]. Indeed, the impairment of Hsp90 function has been shown to enhance the cytotoxicity of a variety of chemical and physical DNA damaging agents (i.e., ionizing radiation (IR)), inhibiting, for instance, ATM-dependent repair mechanism [56,60,61]. In particular, the sensitivity of aneuploid cancer cells to Hsp90 inhibition seems to reflect the increased proteotoxic stress associated to the accumulation of misfolded proteins, as supported by the evidence that Hsp90 inhibition potentiates the effects of proteasome inhibitors in multiple myeloma [62,63]. Among several Hsp90 inhibitors under investigation for cancer treatment, almost all target the N-terminus or the C-terminus of the protein [26]. In particular, inhibitors that bind the N-terminal region containing the ATP pocket represent the most effective anti-cancer drugs (Table 1) [64,65,66,67].
3. Structural Aspects of Hsp90 and Related Chaperones
Hsp90 is a homodimeric protein of ~90 kDa displaying ligand-dependent closed and open conformations. Hsp90 and related chaperones are characterized by three structural domains and two disordered regions: (i) the N-terminal domain contains the ATP binding site and is pivotal for dimerization; (ii) the variable, disordered, and charged region named linker, connecting the N-terminal to the middle domain, participates to nucleotide, co-chaperone, and client protein recognition, and finely modulates the Hsp90 activity acting as a “rheostat”; (iii) the middle domain is involved in the hydrolysis of ATP and participates to co-chaperone recognition; (iv) the C-terminal domain is responsible for Hsp90 dimerization; and (v) the C-terminus unstructured region, ending with the highly conserved Met-Glu-Glu-Val-Asp motif, is involved in the co-chaperone recognition (Figure 1) [29,68].
The structures of the isolated ligand-free and/or ligand-bound N-terminal, middle, and/or C-terminal domains of Escherichia coli Htpg, yeast (Saccharomyces cerevisiae) Hsp90, Plasmodium falciparum Hsp90, Leishmania major Hsp90, Trypanosoma brucei Hsp90, dog Grp94, and human Hsp90 have been determined. The N-terminal domain is a α/β sandwich containing the ATP binding pocket, which extends from the buried face of the anti-parallel β-sheet to the surface (Figure 1). The N-terminal domain of Hsp90 recognizes the natural products geldanamycin and radicicol, inhibiting competitively the hydrolysis of ATP, in a very similar way to ATP. The middle domain of yeast Hsp90 comprises a large α/β/α subdomain at the N-terminus, which is bound to a smaller α/β/α subdomain through a series of short α-helices. A hydrophobic patch, containing a highly conserved solvent exposed Trp residue and an amphipathic loop, which has been postulated to recognize client proteins, characterizes the Hsp90 middle domain. Moreover, the Hsp90 middle domain displays a critical loop, containing a highly conserved Arg residue, which is pivotal for the ATPase activity. The C-terminal domain of E. coli HtpG, displaying a homodimeric structural organization also in the ligand-free protein, is composed by two small mixed α/β domains; the dimerization interface is formed by two α-helices of each subunit which pack together to form a four α-helical bundle (Figure 1) [68].

{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Derivatives | Structure | Pharmacokinetics (nM) | Clinical Study Stage | References |
|---|---|---|---|---|---|
| Geldanamycin (naturally derived from Streptomyces hygroscopicus) |  | GI50 = 1.0 × 10−1 nM; LC50 = 2.1 × 104 nM; Kd = 1.2 × 103 nM | Preclinical | [61,65,69]; www.medchemexpress.net | |
| 17-AAG (17-allylamino-17-desmethoxygeldanamycin; tanespimycin) |  | IC50 = 5.0 × 101–1.0 × 104 nM | Phase I/II/III | [70,71,72,73,74,75,76,77,78]; www.clinicaltrials.gov; www.medchemexpress.net | |
| 17-DMAG (17-dimethylaminoethylamino-17-demetoxygelanamycin; alvespimycin) |  | IC50 = 6.0 × 101–3.0 × 103 nM; Kd = 3.5 × 102 nM | Phase I/II | [72,77,79,80,81]; www.clinicaltrials.gov | |
| PU-H71 |  | IC50 = 5.0 × 101–3.0 × 102 nM | Phase I | [82,83,84]; www.clinicaltrials.gov | |
| NVP-AUY922 |  | IC50 = 1.3 × 101 nM (Hsp90α); IC50 = 2.1 × 101 nM (Hsp90β); GI50 = 2.0–4.0 × 101 nM; Kd = 1.7 nM | Phase I/II | [85,86,87]; www.medchemexpress.net; www.clinicaltrials.gov | |
| Radicicol (naturally derived from Diheterospora chlamydosporia) |  | Kd = 1.9 × 101 nM; IC50 = 2.0 × 101 nM | Preclinical | [65] | |
| STA9090 (Ganetespib) |  | IC50 = 1.0–5.0 × 101 nM | Phase I/II/III | [88,89,90,91]; www.clinicaltrials.gov; www.medchemexpress.net |
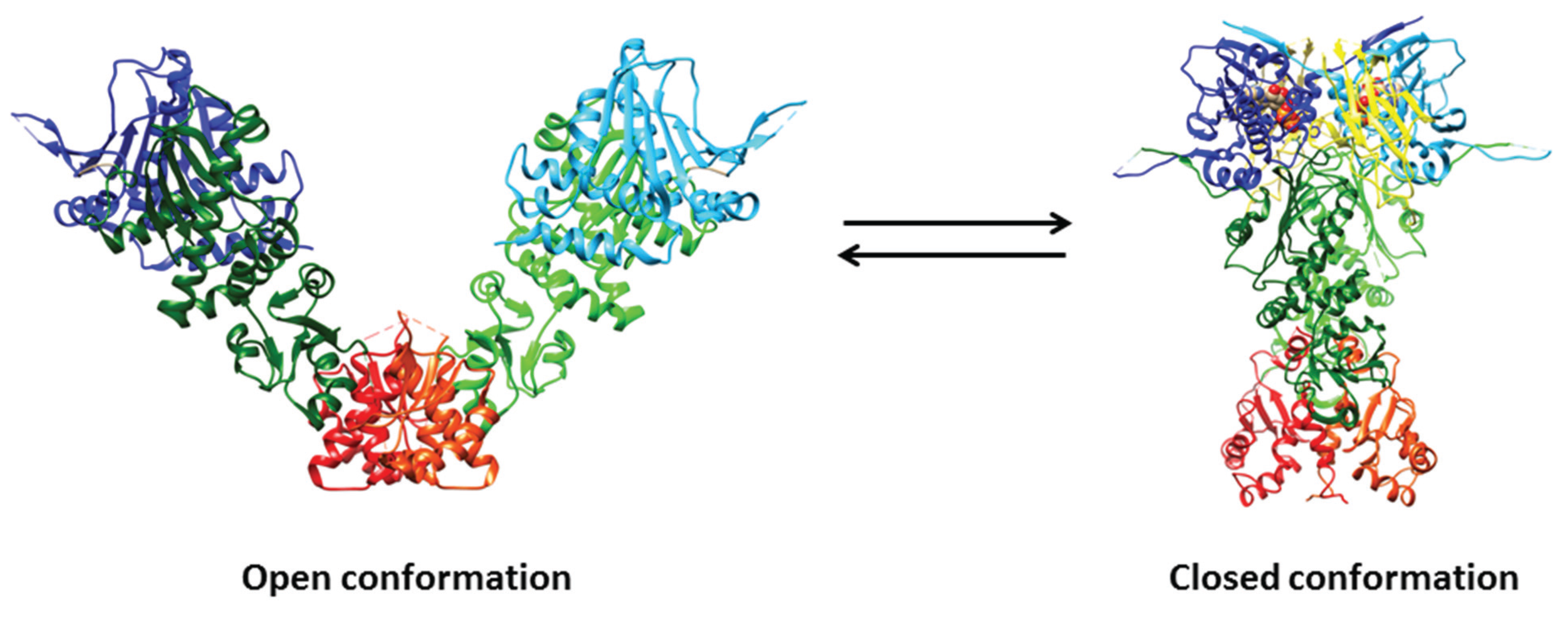
The open (i.e., ligand-free) structure of full-length E. coli HtpG and the closed (i.e., ligand-bound) structures of full-length yeast Hsp90, E. coli HtpG, and dog Grp94 have been determined. Overall, the structures of the three domains of the full-length proteins are very similar to those of the isolated domains; however, some differences were observed in the N-terminal domain (possibly reflecting nucleotide binding and dimerization) and in the α-helical segment that protrudes out from the C-terminal domain (Figure 1) [68].
Figure 1.
Crystal structures of open (i.e., ligand-free) full-length Hsp90 from E. coli (HtpG; PDB ID: 2IOQ) and closed (i.e., ATP- and p23-bound) yeast Hsp90 (PDB ID: 2CG9). The N-domain is depicted in blue and cyan, the middle domain in dark green and light green, and the C-domain in red and orange. The p23 co-chaperone is in yellow, whereas ATP is depicted through a space-filling representation. Pictures were drawn by UCSF-Chimera [92].
Figure 1.
Crystal structures of open (i.e., ligand-free) full-length Hsp90 from E. coli (HtpG; PDB ID: 2IOQ) and closed (i.e., ATP- and p23-bound) yeast Hsp90 (PDB ID: 2CG9). The N-domain is depicted in blue and cyan, the middle domain in dark green and light green, and the C-domain in red and orange. The p23 co-chaperone is in yellow, whereas ATP is depicted through a space-filling representation. Pictures were drawn by UCSF-Chimera [92].

The structure of the open full-length E. coli HtpG displays an open V-like conformation. The solvent-exposed hydrophobic patches appear to represent potential binding sites for client proteins, whose recognition by Hsp90 is favored by its flexible conformation. In the ligand-free full-length E. coli HtpG, the lid of the ATP binding pocket is positioned in such a way as to block nucleotide binding, representing a case of auto-inhibitory mechanism. A large structural rearrangement(s) of the N-terminal domain relative to the middle domain is pivotal to allow the closure of the lid after nucleotide binding [93].
In contrast to the open ligand-free form of full-length E. coli HtpG, the closed form of yeast Hsp90 complexed with the AMP-PNP and p23 is relatively compact (Figure 2) and characterized by: (i) the dimerization not only of the C-terminal regions, but also of the N-terminal domains; (ii) extensive contacts between the domains within each monomer; and (iii) twisting of the subunits. In particular, the ATP binding region undergoes a large movement around a hinge created by two critical Gly residues, thus exposing a hydrophobic patch that contributes to build up the dimerization surface of the N-terminal domains [94]. The two p23 co-chaperones bind into grooves occurring at the interface between the two N-terminal domains of yeast Hsp90, through an ATP-dependent mechanism. Although the inspection of the yeast Hsp90-p23-AMP-PP structure does not directly reveal how p23 inhibits the ATPase activity of Hsp90, it has been postulated that p23 stabilizes the closed dimerized state of full-length Hsp90 preventing the release of ADP and phosphate [94].
Figure 2.
Crystal structure of yeast Hsp90/ATP/p23 closed chaperone complex (PDB ID: 2CG9). The Hsp90 N-terminal domain is shown in cyan, the p23 co-chaperone is in yellow, and ATP is depicted through a space-filling representation. Pictures were drawn by UCSF-Chimera [92].
Figure 2.
Crystal structure of yeast Hsp90/ATP/p23 closed chaperone complex (PDB ID: 2CG9). The Hsp90 N-terminal domain is shown in cyan, the p23 co-chaperone is in yellow, and ATP is depicted through a space-filling representation. Pictures were drawn by UCSF-Chimera [92].

4. Role of Hsp90 in the Genome Stability Maintenance
Cells respond to DNA damage by activating the complex DNA damage response (DDR) pathway that includes the cell cycle arrest, the transcriptional and post-translational activation of a subset of genes including those associated with DNA repair, and, under some circumstances, the triggering of programmed cell death. The efficacy of the DDR is influenced by the nuclear levels of DNA repair proteins, which are regulated by balancing between protein synthesis and degradation, and the control of nuclear import and export. The inability to repair DNA damage leads to genetic instability, which in turn may enhance the rate of cancer development [95,96,97].
Historically, the Hsp90 molecular chaperone system had been viewed as a strict cytosolic machine [17]. Now it is becoming clear that Hsp90 and its associated co-chaperones have important functions into the nucleus, including chromatin remodeling, DNA transcription, RNA processing, DNA replication, telomere maintenance, and DNA repair [15,98]. Indeed, Hsp90 participates to DNA repair and interacts with DNA metabolic proteins via the p23 co-chaperone or upon phosphorylation [15,98,99,100].
4.1. The DNA Double-Strand Break Response
Among several types of lesion, the DNA double-strand break (DSB) is one of the most deleterious and harmful. DSBs arise from both endogenous and exogenous sources, including reactive nitrogen and oxygen species, replication errors, chemical mutagens, and IR [101].
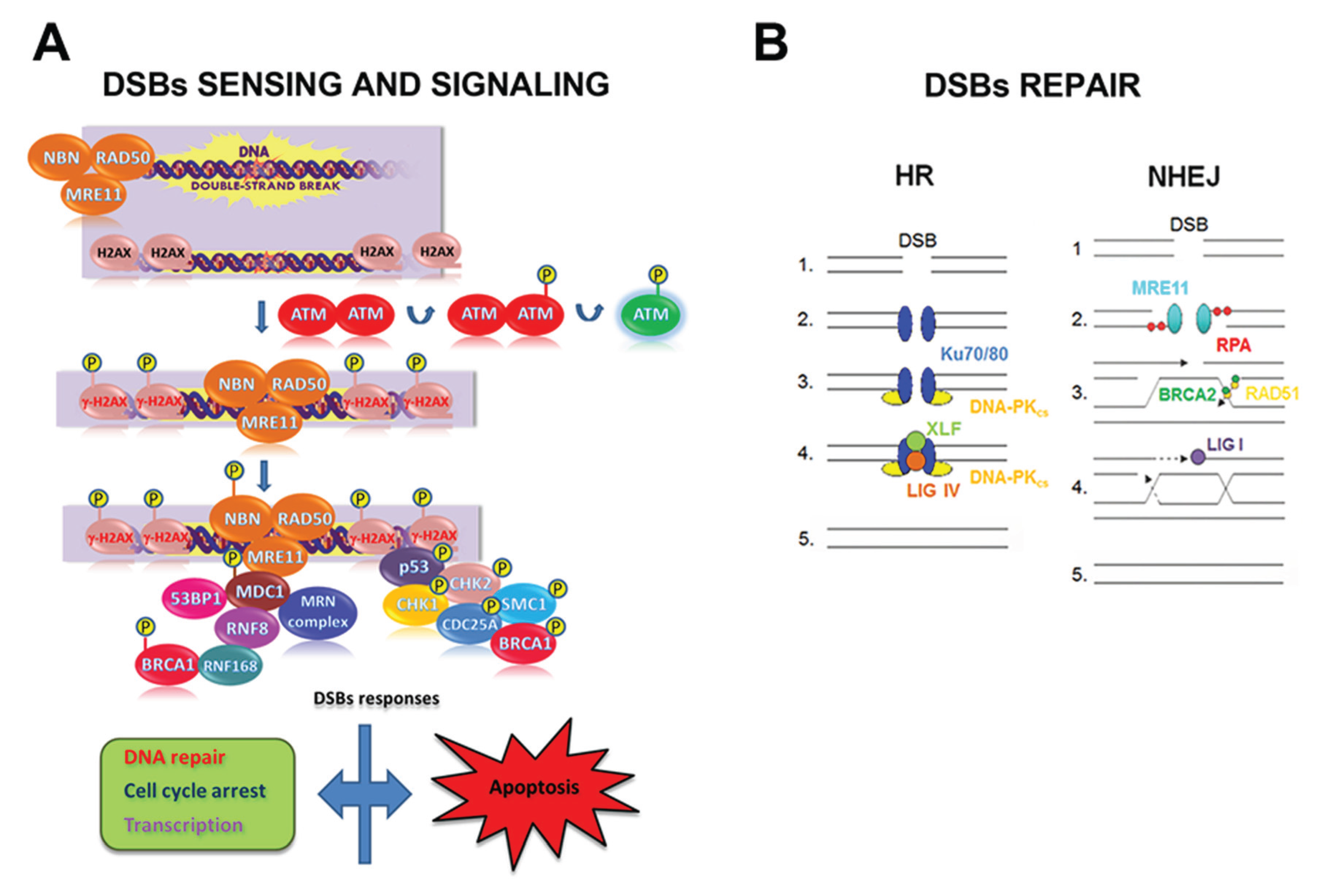
DSB elimination can be divided in three phases (i.e., sensing, signaling, and repair) and involves the spatio-temporal orchestration of a high number of proteins. Briefly, the sensing phase includes: (i) DSB recognition by the MRE11/RAD50/NBN complex; (ii) ATM activation; (iii) histone H2AX phosphorylation (the phosphorylated form of H2AX is named γ-H2AX), (iv) MDC1 recruitment; and (v) MRE11/RAD50/NBN retention (leading to further ATM activation and γ-H2AX spreading). γ-H2AX, the initial DSB sensor, represents the site for the accumulation of signaling and repair proteins to DNA breaks to form the so-called IR-induced foci (IRIF) [102,103]. After sensing the damage, the signaling phase is activated. Indeed, DSBs trigger a myriad of post-translational modifications that alter catalytic activities and specific protein-protein interactions, followed by the reversal of these changes as the repair is completed (Figure 3).
Figure 3.
Double-strand break (DSB)s sensing, signaling and repair in mammalian cells. (A) Schematic representation of the early steps of DSBs sensing and signaling. After a DSB induction: (i) the MRE11/RAD50/NBN complex, made up of MRE11, RAD50 and NBN, localizes at damage site; (ii) ATM undergoes auto-phosphorylation at the Ser1981 residue, with the consequent dissociation of ATM dimers and ATM activation; (iii) ATM phosphorylates H2AX histone that, as γ-H2AX, serves as a platform for the assembly of proteins involved in DNA repair, cell checkpoint response, and transcription. Depending on the severity of the DNA damage and on the cell type, cells may undergo apoptosis; (B) Once the cell has sensed the DSB, the DNA repair machinery is recruited to the lesion in relation to the cell cycle stage. In G1 phase cells undergo repair predominantly through NHEJ repair pathway, whereas in G2/M the presence of replicated DNA allows the repair through the HR pathway. So, during the NHEJ pathway, in the presence of a DSB (1), the broken ends are bound by Ku70/80 heterodimer (2) that recruit DNA-PKCS (3). After the ends have been processed, the XRCC4/Ligase IV complex completes the final ligation step (4) and the damage is repaired (5). On the contrary, in the presence of a DSB (1), the HR pathway requires MRE11 or Exo1 exonuclease activity to resect the DNA ends forming a 3’ overhang; this structure is stabilized by RPA (2) and then loaded on the homologous DSB region by the strand exchange protein RAD51 and by BRCA2 (3), leading to the formation of the Holliday junctions intermediate (4). Endonuclease and resolvase proteins are involved in resolving the Holliday junctons intermediate (5).
Figure 3.
Double-strand break (DSB)s sensing, signaling and repair in mammalian cells. (A) Schematic representation of the early steps of DSBs sensing and signaling. After a DSB induction: (i) the MRE11/RAD50/NBN complex, made up of MRE11, RAD50 and NBN, localizes at damage site; (ii) ATM undergoes auto-phosphorylation at the Ser1981 residue, with the consequent dissociation of ATM dimers and ATM activation; (iii) ATM phosphorylates H2AX histone that, as γ-H2AX, serves as a platform for the assembly of proteins involved in DNA repair, cell checkpoint response, and transcription. Depending on the severity of the DNA damage and on the cell type, cells may undergo apoptosis; (B) Once the cell has sensed the DSB, the DNA repair machinery is recruited to the lesion in relation to the cell cycle stage. In G1 phase cells undergo repair predominantly through NHEJ repair pathway, whereas in G2/M the presence of replicated DNA allows the repair through the HR pathway. So, during the NHEJ pathway, in the presence of a DSB (1), the broken ends are bound by Ku70/80 heterodimer (2) that recruit DNA-PKCS (3). After the ends have been processed, the XRCC4/Ligase IV complex completes the final ligation step (4) and the damage is repaired (5). On the contrary, in the presence of a DSB (1), the HR pathway requires MRE11 or Exo1 exonuclease activity to resect the DNA ends forming a 3’ overhang; this structure is stabilized by RPA (2) and then loaded on the homologous DSB region by the strand exchange protein RAD51 and by BRCA2 (3), leading to the formation of the Holliday junctions intermediate (4). Endonuclease and resolvase proteins are involved in resolving the Holliday junctons intermediate (5).

Two fundamentally different DSB repair pathways, i.e., the non-homologous end-joining (NHEJ) and the homologous recombination (HR), have been identified both in mammalian cells and in yeast [104,105,106,107]. Both NHEJ and HR DSB repair pathways may be activated simultaneously and cooperatively to repair DNA lesions [108,109,110].
NHEJ (i) brings the DNA termini together in a protein-DNA complex; (ii) does not require the presence of an undamaged template; and (iii) is active throughout the whole cell cycle [111,112]. Therefore, NHEJ is considered as a flexible but conservative DNA repair mechanism that enables a direct rejoining of broken DNA termini, although through an error-prone process [108,113]. To initiate NHEJ, the Ku70/80 heterodimer binds to blunt or near-blunt DNA ends. The DSB-Ku70/80 heterodimer complex recruits and activates the DNA-PKcs adduct that triggers an extensive signaling cascade orchestrating downstream repair processes [114]. NHEJ repair is facilitated by the scaffold proteins XRCC4 and XLF (also called Cernunnos) that bind DNA Ligase IV, the enzyme responsible for sealing the break. If DNA ends need nucleolytic processing before ligation, the Artemis endonuclease, a DNA-PKcs-interacting protein, provides this activity (Figure 3) [111,115].
The HR mechanism depends on the use of a template, as can be found on a sister chromatid during the S and G2 cell cycle phase. Since the HR mechanism uses an undamaged DNA template to restore chromosome integrity, it has the potential to repair DSBs more faithfully than NHEJ. The search for sequence homology to repair DNA requires the presence of single-strand DNA at the DSB end [97,111,116]. This intermediate can be generated by the nucleolytic degradation of the 5’ strand of a DSB end in a process mediated by the MRE11/RAD50/NBN complex. This complex recruits CtIP and initiates the resection, the EXO1 and DNA2 nucleases perform the bulk of end-resection required for HR. In this process, DNA2 acts in complex with the RecQ helicases BLM and/or WRN (Figure 3) [117].
In mammalian cells, the HR mechanism seems to be less utilized than NHEJ in the repair of DSBs, but defects in the HR mechanism do enhance cellular radio-sensitivity. Since most cells arrest at the G2 phase in response to IR exposure and the HR mechanism mainly works in late-S and G2 phases, inhibiting the HR mechanism would be a good strategy for cancer cells treatment [118].
4.2. Hsp90 and the DNA Damage Response Clients
Although the cellular circumstances that incorporate Hsp90 into an optimal DNA damage response to IR remains still unknown, multiple components of the DSBs repair machinery, including BRCA1, BRCA2, RAD51, CHK1, DNA-PKcs, members of the FA pathway, histones, and components of the MRE11/RAD50/NBN complex have been reported to be Hsp90 clients [60,119,120,121,122,123,124,125]. Of note, the Hsp90 inhibitors geldanamycin (a highly cytotoxic natural antibiotic isolated from Streptomyces hygroscopicus, which binds Hsp90 in the ATP-binding site) [4,65,69,126], its derivates 17-(allylamino)-17-demethoxygeldanamycin (17-AAG) [26,70,127] and 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG) [59,71,72,79,127,128], PU-H71 (a member of a class of inhibitors with purine scaffold) [129,130], NVP-AUY922 (a member of a class of inhibitors possessing a pyrazole scaffold) [85,127,131,132], ganetespib (also named STA-9090) [88], and radicicol [65,133] affect the DNA repair, thus highlighting the pivotal role of Hsp90 in the DDR (Table 1) [25,26,57,60,134,135,136].
4.2.1. BRCA1, BRCA2, and RAD51
BRCA1 is a nuclear tumor suppressor critical for DSBs and inter-strand cross-links by the HR mechanism [137]. BRCA1 is phosphorylated by ATM, ATR, and CHK2 kinases in response to DNA damage, then phosphorylated BRCA1 organizes multiple distinct protein complexes that recognize and repair damaged DNA and activate cell cycle checkpoints [138,139]. Except for DNA-PKcs, the activity and/or recruitment of BRCA2, CHK1, FANCA, the MRE11/RAD50/NBN complex, and RAD51 to the DSBs is BRCA-dependent [139,140,141,142,143].
Among the proteins involved in the HR mechanism and sensitive to Hsp90 inhibitors, BRCA1 appears to be the most upstream, being sensitive to the Hsp90 inhibitor 17-AAG [124]. Since tumor cells expressing high levels of BRCA1 are resistant to both IR and several classes of chemotherapeutic agents, the ablation of BRCA1 expression through Hsp90 inhibitors may restore sensitivity to anti-tumor agents, thus representing a possible cancer therapeutic strategy [124,144]. Moreover, the intrinsic sensitivity of BRCA1-mutant or -deficient cells to 17-AAG suggests that this agent might also show efficacy in both primary BRCA1 mutant tumors as well as in sporadic tumors that have lost BRCA1 expression by non-mutational means [124].
BRCA2 directly interacts with Hsp90, 17-AAG causing BRCA2 degradation [57]. Since BRCA2 promotes RAD51-mediated HR at sites of DSBs [145], 17-AAG-dependent BRCA2 degradation leads to RAD51 impairment and induces a delay in the IR-induced BRCA2-mediated RAD51 foci formation. After irradiation, RAD51 foci formation is also inhibited by the treatment of cancer cells with PU-H71 and NVP-AUY922 [136,146]. Both non-irradiated and irradiated cells treated with ganetespib are characterized by the degradation of RAD51 and by a lowered activation of the ATM kinase and other proteins involved in DNA repair [89]. The delay in RAD51 focal assembly leads to a decrease in the rate of the successful HR mechanism. In turn, the persistence of unrepaired DNA damage may increase the time over which cells trigger apoptosis because of DSBs. Moreover, the persistence of unrepaired DNA damage combined with CHK1 depletion (see paragraph 4.2.2) is also likely to increase the probability of an attempted cell cycle progression with an unacceptable/unsafe burden of DNA damage [136].
4.2.2. CHK1
The DNA damage-activated cell cycle checkpoint pathways are evolutionarily conserved signaling pathways that regulate cell cycle progression, programmed cell death, and DNA repair [149]. CHK1 is a key regulator of the signaling pathway activated by DNA replication stress and DNA damage. Indeed, following DNA replication fork stalling, single-stranded regions of DNA (ssDNA) accumulate, RPA binds to ssDNA, and the ATR kinase localizes at the stalled fork together with its binding partner ATRIP [150,151]. The stalled fork also recruits DNA polymerase α, which then participates in the RAD17-dependent recruitment of the PCNA-like RAD9-Hus1-RAD1 clamp complex to chromatin. Once bound to chromatin, the RAD9-Hus1-RAD1 complex facilitates the ATR-mediated phosphorylation and activation of CHK1 [152].
CHK1 performs several functions that support cell survival. Indeed, CHK1 (i) promotes cells arrest in G2; (ii) slows the progression through the S phase by inhibiting the initiation of new replication sites and stimulating the firing of dormant origins placed in proximity of stalled forks; and (iii) avoids fork collapse into DSBs. At the molecular level, CHK1 is activated at stalled replication forks, the nucleoplasm representing the site where CHK1 phosphorylates target proteins such as Cdc25C and Cdc25A, two cell cycle phosphatases that in turn activate the Cdk1-cyclin B complex. Because Cdc25A is also required for the activation of Cdk2 complexes, which then control the firing of origins of replication, the activation of this pathway blocks the S phase progression [152]. Correspondingly, the disruption of the CHK1 signaling pathway is associated with increased sensitivity to genotoxins [153]. Additionally, cells in which CHK1 has been deleted by gene targeting are sensitive to the replication inhibitor aphidicolin and IR [154].
CHK1 is an Hsp90 client, indeed the 17-AAG Hsp90 inhibitor leads to CHK1 degradation and to the disruption of the CHK1-mediated Cdc25A degradation. Overall, Hsp90-dependent modulation of CHK1 stability highlights a critical role played by this kinase in maintaining cell viability following replication stress. Of note, the disruption of CHK1 function(s) by 17-AAG sensitize tumor cells arrested in the S phase by gemcitabine treatment. This suggests that the blockage of CHK1 signaling enhances the efficacy of chemotherapic agents [119]. NVP-AUY922 also reduces the CHK1 levels to the sites of DNA damage in a dose-dependent manner, suppressing the CHK1-mediated G2 arrest in the presence of IR-induced DSBs [136].
4.2.3. DNA-PK
The catalytic subunit of DNA-PK is a client of Hsp90 in the epithelioid cervix carcinoma cell line (HeLa), but not in normal the embryonic kidney human cell line (HEK293). Moreover, the cytosolic but not the nuclear DNA-PK levels are reduced in HeLa cells after treatment with the Hsp90 inhibitor radicicol [135]. The IR-induced activation of DNA-PK involves the interaction with ErbB1, which mediates DNA-PK transport into the nucleus [155]. Of note, human epithelial cells derived from pancreas carcinoma (MiaPaCa) exposed to 17-DMAG show the reduction of (i) the ErbB1 activity; (ii) the IR-induced interaction between ErbB1 and DNA-PK; and (iii) the IR-induced activation of DNA-PK, compromising the DSBs repair [60].
Hsp90α is phosphorylated extensively by DNA-PK at Thr5 and Thr7 in the nucleus of cells engaged to apoptosis. Phosphorylation is part of a signaling pathway activated by the TRAIL-induced apoptosis, by the Fas ligand, and by the apoptosis-inducer staurosporine [156,157]. An epigenetic landmark of early apoptosis is H2AX phosphorylation at the nuclear periphery of cells committed to apoptosis. This γ-H2AX nuclear staining is termed “the apoptotic γ-H2AX ring” that, although morphological distinct from the DDR, is characterized by the presence of several phosphorylated DDR proteins, such as ATM, NBN, CHK2, and DNA-PK [158,159]. Notably, the Thr5 and Thr7 phosphorylated form of Hsp90α also localizes into the apoptotic ring, where it acts as a modulator of the apoptotic process, enables the stabilization and activation of DNA-PK, and allows the execution of nuclear apoptosis [99,100]. Remarkably, Hsp90 inhibitors such as geldanamycin and 17-AAG enhance TRAIL-induced DNA-PK and H2AX activation [100,160].
Although both the DDR and the apoptotic ring are mediated by DNA-PK, the apoptosis-induced phosphorylation of Hsp90α at Thr5 and Thr7 residues differs from that induced from DDR. Indeed, the levels of the apoptosis-induced Hsp90α phosphorylation are markedly greater than those generated by DNA damage inducers [121]. Moreover, the apoptosis-induced Hsp90α phosphorylated form is distributed broadly and rapidly into the apoptotic ring, contrary to the much weaker, focally distributed, and delayed phosphorylation of Hsp90α in γ-H2AX foci formed during the DDR [99]. Finally, the apoptotic ring, unlike DDR foci, is defective for DNA repair because it lacks many DDR effector proteins, such as MDC1 and 53BP1. This is due to the fact that the caspase-mediated cleavage of MDC1, which normally binds to γ-H2AX and initiates the DDR, dissociates the FHA and BRCT domains of MDC1 and inactivates DNA repair in the apoptotic ring [158,161].
4.2.4. The FA Pathway
The nuclear Fanconi Anemia (FA) “core complex” is involved in the FA pathway, also termed FA/BRCA pathway. This complex is composed of eight proteins including FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, and FANCM. FANCD2 and its paralogue/ binding-partner FANCI are activated by the ATR-mediated phosphorylation and by the FA core complex-dependent mono-ubiquitination [123]. Thus, the active forms of FANCD2 and FANCI localize to the chromatin at sites of DNA damage, where they interact with DNA repair proteins including BRCA1 and BRCA2. Indeed, the core complex and FANCD2 contribute in the maintenance of genome stability through the coordination of multiple DNA repair mechanisms, including HR, NHEJ, and translesion synthesis (TLS) [162,163].
Hsp90 regulates the FA pathway. Cell treatment with 17-AAG induces the rapid proteasomal degradation and cytoplasmic retention of FANCA, strongly suggesting that inhibition of Hsp90 induces nuclear depletion of the FA core complex impairing the activation of the FA pathway. Moreover, 17-AAG inhibits the DNA damage-induced activation of FANCD2, enhancing DNA cross-linker-induced cytotoxicity and chromosome aberrations [164,165].
4.2.5. Histones
Changes in chromatin adjacent to the DNA break sites are instrumental to the DDR because they influence chromatin compaction and the binding of repair and signaling proteins to the DNA lesion [166]. Hsp90 interacts with high affinity with all histones; in particular, Hsp90 binding to the C-terminal tail of histone Hl influences chromatin remodeling, which represents a fundamental event in the DSB repair pathways [167]. Indeed, Hsp90 interferes with the regulatory Ser-Pro-Lys-Lys motifs of histone H1 impairing phosphorylation and acetylation during stress or steroid action(s) [168,169,170,171].
Of note, histone H1 has the unique feature to increase the Hsp90-associated Mg2+-dependent kinase activity 6- to 7-fold, which is not shared by other histones despite their similar high affinity for Hsp90. Furthermore, histone H1 has been reported to possess an ATP-binding activity regulating the ATP-turnover of Hsp90 [122].
4.2.6. The MRE11/RAD50/NBN Complex
The treatment of MiaPaCa cells with 17-DMAG abrogates the IR-induced activation of the G2- and S-phase checkpoints, as well as DSB repair [54,60]. These effects seem to be the result of the disruption of the NBN/ATM axis, which in turn leads to a diminished activation of ATM after IR. Although both the levels of phosphorylated ATM and the number of phospho-ATM-containing nuclear foci were reduced in MiaPaCa cells after IR-induced damage, no interaction between ATM and Hsp90 has been detected. Although 17-DMAG neither induces degradation of any of the MRE11/RAD50/NBN components nor disrupts the MRE11/RAD50/NBN complex, nuclear Hsp90 binds the MRE11/RAD50/NBN complex. However, the exposure of MiaPaca cells to 17-DMAG compromises the ability of the MRE11/RAD50/NBN components to form nuclear foci and diminishes the interaction between NBN and ATM [60].
4.3. Other DNA Damage Response Clients
4.3.1. MSH2
The DNA mismatch repair (MMR) pathway maintains genomic integrity by correcting DNA replication errors. MSH2 is a crucial protein involved in the highly conserved MMR mechanism, playing a pivotal role in the recognition of DNA mismatches and in the recruitment of other DNA repair proteins to the mismatched sites [172]. Under oxidative stress, both the p38 MAPK and c-Jun N-terminal kinase (JNK) pathways mediate the ectopic expression of MSH2 in renal carcinoma cell [173,174].
Anti-folates cause DSBs in human non-small-cell lung cancer (NSCLC) [175] and in human colon cancer cells [176]. The anti-folate pemetrexed possesses single-agent activity in front- and second-line treatments of NSCLC, especially adenocarcinoma. Pemetrexed causes a growth and survival response through the EGFR-mediated activation of the PI3K/AKT pathway. The combined treatment of NSCLC and lung adenocarcinoma cells with pemetrexed and with the 17-AAG Hsp90 inhibitor results in an enhanced pemetrexed-induced cytotoxic effect. In particular, the pemetrexed-induced down-regulation of the MKK3/6-p38 MAPK signal causes the reduction of MSH2 levels [174].
Tamoxifen, an estrogen receptor antagonist, has been used as therapy against breast cancer [177] and could promote cell death in NSCLC [178]. The combined treatment of tamoxifen with 17-AAG promotes the cytotoxic effect and growth inhibition of tamoxifen, significantly decreasing the expression of MSH2 in human lung carcinoma cells treated with tamoxifen [179,180].
4.3.2. PCNA and Polymerase η
TLS represents an essential mechanism for bypassing the block in the progress of replication forks. TLS is catalyzed by specialized polymerases, including the Y family polymerases. However, Y-polymerases display low stringency of the active site and a lack of proofreading, thus contributing to mutagenesis. Mammals have four members of Y-polymerases, i.e., Pol η, Pol κ, Pol ι, and REV1 [181,182,183].
UV irradiation causes several types of DNA lesions, including cyclobutane pyrimidine dimers (CPDs) and (6–4) photoproducts ((6–4)PPs). Whereas (6–4)PPs are efficiently removed by the nucleotide excision repair pathway, CPDs frequently escape this mechanism. DNA polymerase η (Pol η) is a member of the mammalian Y family polymerases and performs error-free TLS across CPDs by incorporating correct bases on the opposite strand [184,185]. For this function, Pol η accumulates in nuclear foci at replication stalling sites via its interaction with monoubiquitinated PCNA [186,187]. The molecular chaperone Hsp90 promotes UV irradiation-induced nuclear focus formation of Pol η through control of its stability and binding to monoubiquitinated PCNA. Hsp90 facilitates the folding of Pol η into an active form in which PCNA- and ubiquitin-binding regions are functional. Furthermore, Hsp90 inhibition potentiates UV-induced cytotoxicity and mutagenesis in a Pol η-dependent manner, thus suggesting a role of Hsp90 as an essential regulator of Pol η-mediated TLS [188]. Of note, PCNA forms a complex with Hsp90 in HCT-116 (human colon cancer cell line), HT-29 (human colorectal adenocarcinoma), and in HNSCC-1483 (human head and neck cancer cell line), the treatment with geldanamycin resulting in the degradation of both PCNA and Hsp90 [189].
4.3.3. XRCC1
Base modifications are perhaps the most common type of endogenous DNA damage, accounting for thousands of lesions per mammalian genome per day [190]. DNA alterations encompass alkylative and oxidative base products, abasic sites, strand breaks, and misincorporated nucleotides. The base excision repair (BER) pathway evolved to cope with the high level of spontaneous decay products that are formed in DNA, as well as those damages created upon reactions with natural endogenous chemicals, most notably reactive oxygen species. BER predominantly deals with non-bulky small nucleobase lesions, excising and replacing incorrect (e.g., uracil) or damaged bases derived from deamination, alkylation or oxidation (e.g., 3-methyladenine, 8-oxoG). The BER pathways can be divided in the following steps: (i) recognition and removal of an incorrect or damaged substrate base by a DNA glycosylase to create an abasic site intermediate; (ii) abasic site incision by an apurinic/apyrimidinic endonuclease or apurinic/apyrimidinic lyase; (iii) removal of the remaining sugar fragment by a lyase or phosphodiesterase; (iv) gap filling by a DNA polymerase; and (v) sealing of the nick by a DNA ligase [191]. In particular, upon damage, PARP1 activation triggers BER protein recruitment to facilitate short-patch or long-patch BER via DNA polymerase β (Polβ)-dependent or Polβ-independent mechanisms [192]. The heterodimer formed by XRCC1 and Polβ is necessary for the XRCC1-dependent recruitment of Polβ to the DNA damaged sites [193,194]. Overall, XRCC1 is an important scaffold protein that interacts with PARP1, DNA ligase III and Polβ to facilitate efficient repair of DNA single-strand breaks (SSBs) [195]. If SSBs are not properly repaired, they may be converted into DSBs during DNA replication, eventually leading to genetic instability and tumorigenesis [196]. Furthermore, down-regulation of XRCC1 expression in human breast cancer cell lines resulted in decreased SSB repair capacity and hypersensitivity to methyl methanesulfonate (MMS) [197]. PI3K/AKT pathway regulates the basal expression of XRCC1 in non-irradiated cells, and MKK1/2-ERK1/2 is essential for regulating induction of XRCC1 after radiation exposure [198].
Hsp90 protects the Polβ-unbound XRCC1 from degradation in a Polβ-binding-dependent manner [120]. By binding and stabilizing XRCC1, the phosphorylated form of Hsp90 promotes the formation of additional XRCC1 complexes. Indeed, in the absence of Hsp90 or following Hsp90 inhibition, free XRCC1 is removed by ubiquitin-mediated degradation, although also an undetermined ubiquitin-independent mechanism of XRCC1 degradation has been suggested [120].
Due to the varied and different DNA lesions induced by agents such as IR, H2O2 or cisplatin, the cellular response would likely require XRCC1-mediated repair that is both Polβ-dependent and/or Polβ-independent. Indeed, XRCC1 availability and involvement is crucial in BER sub-pathways but also in other repair processes such as NHEJ or nucleotide excision repair (NER) [199,200]. To date it has not been determined the mechanism through which cells regulate the Polβ/XRCC1 heterodimer nor the role for the Polβ-independent XRCC1. It has been suggested that the repair protein complex architecture for the regulation of such DNA repair processes, and in particular of the BER sub-pathway choice, depends upon the archetype BER complex (Polβ/XRCC1), the stabilized XRCC1/(phospho)-Hsp90 complex and XRCC1. Consistent with this model, complex formation (Polβ/XRCC1 versus XRCC1/Hsp90) varies in response to DNA damage or cell proliferation status. In particular, the XRCC1/Hsp90 or XRCC1/phospho-Hsp90 complexes might promote a role for XRCC1 in the DSB repair [199]. Therefore, the phospho-Hsp90- and proteasome-mediated regulation of Polβ and XRCC1 represent a mechanism of the DNA repair pathway choice that is likely to respond to cell type, cell cycle and exogenous stimuli, shedding light on Hsp90 as a novel functional component of BER that may facilitate BER sub-pathway choice in response to DNA damage and cellular proliferation [120].
Gefitinib is a selective epidermal growth factor receptor-tyrosine kinase inhibitor (EGFR-TKI) that blocks growth factor-mediated cell proliferation and ERK1/2 and AKT signaling activation. Furthermore, 17-AAG down-regulated the phospho-ERK1/2, phospho-AKT, and XRCC1 protein levels in gefitinib-exposed NSCLC cells. In addition, 17-AAG improved cell growth inhibition of gefitinib and potently reduced cell viability in both gefitinib-treated A549 and H1975 cell lines. These data provide a strong rationale for using an Hsp90 inhibitor in combination with EGFR-targeted therapies for the treatment of NSCLC [201].
5. Conclusions and Perspectives
Adaptation to stress is not only the driving force for species evolution but also a daily challenge that cells and organisms struggle against during their individual existence. An important mechanism for coping with stresses is the induction of molecular chaperones. These proteins, such as Hsp90, assist protein folding and protein complex assembly and act on misfolded proteins induced by stress conditions such as high levels of reactive oxygen species. Chaperone induction is a mode of adaption that does not involve genetic changes but rather reflects an evolved capacity to maintain proteostasis. The key role played by Hsp90 in the DNA damage sensing signaling, and repair, supported by the number of clients having a role in the DDR, leads to the hypothesis that genome instability arises when the demand for Hsp90 exceeds its functional capacity. Indeed, significant advances have been made recently in the elucidation of the molecular mechanisms of the DSBs damage response to IR, carcinogens, and environmental factors. The importance of a robust DNA-damage surveillance network is evidenced by the fact that defects in sensing, signaling and repair of DNA damage are linked to the development of inherited chromosome instability syndromes and cancer onset. The possibility to specifically interfere with the DSBs repair mechanisms for the development of novel therapeutics may have a significant clinical impact for the development of highly specific cancer therapies.
Cancer cells, which are presumed to be under higher stress conditions than normal cells, are characterized by increased Hsp90 chaperone expression. Furthermore, since Hsp90 and many co-chaperones undergo post-translational modifications upon cell stress, this may lead to biased assembly of Hsp90 complexes targeted to a subset of clients. The preferential chaperoning of some proteins over others could allow the Hsp90 machinery to restore homeostasis in a way tailored to a particular stress. However, a change in specificity implies that certain clients may no longer be chaperoned, thus causing genome instability.
Understanding the role of Hsp90 in genome stability has important implications for human disease. Although the susceptibility to disease may be genetically determined, a reasonable precept is that the actual transition to the disease state at the cellular level occurs when the assembly and plasticity of the encoded protein networks are compromised. Therefore, the primary function of the Hsp90 chaperone machinery may be to buffer against such transitions and, consequently, to disease. In light of these hypotheses, is reasonable to raise concerns about a therapeutic approach based solely on Hsp90 inhibition initially slowing tumor growth and ultimately favoring adaptation to chemotherapy. An alternative approach based on the simultaneous inhibition of Hsp90 and of proteasome-dependent protein degradation has been proposed. This may cause the complete loss-of-function and fast cell death, or simultaneously inhibiting Hsp90 and mitosis to prevent the emergence of chromosomally abnormal cell progeny. However, such drastic treatments are likely to increase unintended side effects on patients. The better understanding of Hsp90 client specificity and regulation under stress may enable more targeted approaches to prevent cancer cell proliferation as well as the potential for adaptive mutations.
Acknowledgments
This work was partially supported by grants from the Italian Ministry of Education, University and Research (CAL 2015 to Alessandra di Masi).
Author Contributions
Rosa Pennisi, Paolo Ascenzi and Alessandra di Masi analyzed data in literature and wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 17-AAG | 17-(allylamino)-17-demethoxygeldanamycin |
| 17-DMAG | 17-dimethylaminoethylamino-17-demethoxygeldanamycin |
| BER | base excision repair |
| DDR | DNA damage response |
| DSB | DNA double-strand break |
| FA | Fanconi anemia |
| HR | homologous recombination repair |
| Hsp90 | heat shock protein 90 |
| IR | ionizing radiation |
| IRIF | IR-induced foci |
| MMR | DNA mismatch repair |
| NHEJ | non-homologous end-joining repair |
| NSCLC | non-small-cell lung cancer |
| Pol η | polymerase η |
| ssDNA | single-stranded regions of DNA |
| SSB | DNA single-strand break |
| TLS | translesion synthesis |
References
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Makhnevych, T.; Houry, W.A. The role of Hsp90 in protein complex assembly. Biochim. Biophys. Acta 2012, 1823, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Neckers, L. Heat shock protein 90: The cancer chaperone. J. Biosci. 2007, 32, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Wandinger, S.K.; Richter, K.; Buchner, J. The Hsp90 chaperone machinery. J. Biol. Chem. 2008, 283, 18473–18477. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Davey, M.; Hsu, Y.C.; Kaplanek, P.; Tong, A.; Parsons, A.B.; Krogan, N.; Cagney, G.; Mai, D.; Greenblatt, J.; et al. Navigating the chaperone network: An integrative map of physical and genetic interactions mediated by the Hsp90 chaperone. Cell 2005, 120, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Schnaider, T.; Soti, C.; Prohaszka, Z.; Nardai, G. The 90-kDa molecular chaperone family: Structure, function, and clinical applications. A comprehensive review. Pharmacol. Ther. 1998, 79, 129–168. [Google Scholar] [CrossRef]
- Sreedhar, A.S.; Kalmar, E.; Csermely, P.; Shen, Y.F. Hsp90 isoforms: Functions, expression and clinical importance. FEBS Lett. 2004, 562, 11–15. [Google Scholar] [CrossRef]
- Echtenkamp, F.J.; Freeman, B.C. Expanding the cellular molecular chaperone network through the ubiquitous cochaperones. Biochim. Biophys. Acta 2012, 1823, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Quadroni, M.; Potts, A.; Waridel, P. Hsp90 inhibition induces both protein-specific and global changes in the ubiquitinome. J. Proteom. 2015, 29, 120–215. [Google Scholar] [CrossRef] [PubMed]
- Young, J.C.; Moarefi, I.; Hartl, F.U. Hsp90: A specialized but essential protein-folding tool. J. Cell Biol. 2001, 154, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Catlett, M.G.; Kaplan, K.B. Sgt1p is a unique co-chaperone that acts as a client adaptor to link Hsp90 to Skp1p. J. Biol. Chem. 2006, 281, 33739–33748. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Rutherford, S.L.; Miyata, Y.; Yahara, I.; Freeman, B.C.; Yue, L.; Morimoto, R.I.; Lindquist, S. Cdc37 is a molecular chaperone with specific functions in signal transduction. Genes Dev. 1997, 11, 1775–1785. [Google Scholar] [CrossRef] [PubMed]
- Echtenkamp, F.J.; Zelin, E.; Oxelmark, E.; Woo, J.I.; Andrews, B.J.; Garabedian, M.; Freeman, B.C. Global functional map of the p23 molecular chaperone reveals an extensive cellular network. Mol. Cell 2011, 43, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Soroka, J.; Wandinger, S.K.; Mausbacher, N.; Schreiber, T.; Richter, K.; Daub, H.; Buchner, J. Conformational switching of the molecular chaperone Hsp90 via regulated phosphorylation. Mol. Cell 2012, 45, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Pratt, W.B.; Dittmar, K.D. Studies with Purified Chaperones Advance the Understanding of the Mechanism of Glucocorticoid Receptor-hsp90 Heterocomplex Assembly. Trends Endocrinol. Metab. 1998, 9, 244–252. [Google Scholar] [CrossRef]
- Workman, P. Altered states: Selectively drugging the Hsp90 cancer chaperone. Trends Mol. Med. 2004, 10, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Iannotti, A.M.; Rabideau, D.A.; Dougherty, J.J. Characterization of purified avian 90,000-Da heat shock protein. Arch. Biochem. Biophys. 1988, 264, 54–60. [Google Scholar] [CrossRef]
- Lees-Miller, S.P.; Anderson, C.W. The human double-stranded DNA-activated protein kinase phosphorylates the 90-kDa heat-shock protein, hsp90 alpha at two NH2-terminal threonine residues. J. Biol. Chem. 1989, 264, 17275–17280. [Google Scholar] [PubMed]
- Welch, W.J.; Feramisco, J.R. Purification of the major mammalian heat shock proteins. J. Biol. Chem. 1982, 257, 14949–14959. [Google Scholar] [PubMed]
- Welch, W.J. The role of heat-shock proteins as molecular chaperones. Curr. Opin. Cell Biol. 1991, 3, 1033–1038. [Google Scholar] [CrossRef]
- Jhaveri, K.; Ochiana, S.O.; Dunphy, M.P.; Gerecitano, J.F.; Corben, A.D.; Peter, R.I.; Janjigian, Y.Y.; Gomes-DaGama, E.M.; Koren, J., III; Modi, S.; et al. Heat shock protein 90 inhibitors in the treatment of cancer: Current status and future directions. Expert. Opin. Investig. Drugs 2014, 23, 611–628. [Google Scholar] [CrossRef] [PubMed]
- Proia, D.A.; Kaufmann, G.F. Targeting heat-shock protein 90 (Hsp90) as a complementary strategy to immune checkpoint blockade for cancer therapy. Cancer Immunol. Res. 2015, 3, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Tummalapalli, S.R.; Rotella, D.P. Progress in the discovery and development of heat shock protein 90 (Hsp90) inhibitors. J. Med. Chem. 2014, 57, 8718–8728. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, K.; Taldone, T.; Modi, S.; Chiosis, G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim. Biophys. Acta 2012, 1823, 742–755. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.S. The Hsp90 chaperone machinery: From structure to drug development. BMB. Rep. 2009, 42, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Banerji, U.; Tavana, B.; George, G.C.; Aaron, J.; Kurzrock, R. Targeting the molecular chaperone heat shock protein 90 (HSP90): Lessons learned and future directions. Cancer Treat. Rev. 2013, 39, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Buchner, J. Structure, function and regulation of the Hsp90 machinery. Biomed. J. 2013, 36, 106–117. [Google Scholar] [PubMed]
- Lianos, G.D.; Alexiou, G.A.; Mangano, A.; Mangano, A.; Rausei, S.; Boni, L.; Dionigi, G.; Roukos, D.H. The role of heat shock proteins in cancer. Cancer Lett. 2015, 360, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Rodina, A.; Chiosis, G. Heat shock protein 90: Translation from cancer to Alzheimer’s disease treatment? BMC Neurosci. 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Mahanta, S. Association of heat-shock proteins in various neurodegenerative disorders: Is it a master key to open the therapeutic door? Mol. Cell. Biochem. 2014, 386, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Yeyati, P.L.; van Heyningen, V. Incapacitating the evolutionary capacitor: Hsp90 modulation of disease. Curr. Opin. Genet. Dev. 2008, 18, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Camphausen, K.; Tofilon, P.J. Inhibition of Hsp90: A multitarget approach to radiosensitization. Clin. Cancer Res. 2007, 13, 4326–4330. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Thao, L.; Sensintaffar, J.; Zhang, L.; Boehm, M.F.; Fritz, L.C.; Burrows, F.J. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 2003, 425, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Kirschke, E.; Goswami, D.; Southworth, D.; Griffin, P.R.; Agard, D.A. Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell 2014, 157, 1685–1697. [Google Scholar] [CrossRef] [PubMed]
- Neckers, L.; Ivy, S.P. Heat shock protein 90. Curr. Opin. Oncol. 2003, 15, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Sangster, T.A.; Lindquist, S.; Queitsch, C. Under cover: Causes, effects and implications of Hsp90-mediated genetic capacitance. Bioessays 2004, 26, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Segnitz, B.; Gehring, U. The function of steroid hormone receptors is inhibited by the hsp90-specific compound geldanamycin. J. Biol. Chem. 1997, 272, 18694–18701. [Google Scholar] [CrossRef] [PubMed]
- Sharp, S.; Workman, P. Inhibitors of the HSP90 molecular chaperone: Current status. Adv. Cancer Res. 2006, 95, 323–348. [Google Scholar] [PubMed]
- Neckers, L.; Workman, P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Chiosis, G.; Neckers, L. Tumor selectivity of Hsp90 inhibitors: The explanation remains elusive. ACS Chem. Biol. 2006, 1, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Ferrarini, M.; Heltai, S.; Zocchi, M.R.; Rugarli, C. Unusual expression and localization of heat-shock proteins in human tumor cells. Int. J. Cancer 1992, 51, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K.; Khaleque, M.A.; Sawyer, D.B.; Ciocca, D.R. Heat shock proteins in cancer: Chaperones of tumorigenesis. Trends Biochem. Sci. 2006, 31, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Chiosis, G.; Vilenchik, M.; Kim, J.; Solit, D. Hsp90: The vulnerable chaperone. Drug Discov. Today 2004, 9, 881–888. [Google Scholar] [CrossRef]
- Fionda, C.; Soriani, A.; Malgarini, G.; Iannitto, M.L.; Santoni, A.; Cippitelli, M. Heat shock protein-90 inhibitors increase MHC class I-related chain A and B ligand expression on multiple myeloma cells and their ability to trigger NK cell degranulation. J. Immunol. 2009, 183, 4385–4394. [Google Scholar] [CrossRef] [PubMed]
- Alexiou, G.A.; Vartholomatos, G.; Stefanaki, K.; Patereli, A.; Dova, L.; Karamoutsios, A.; Lallas, G.; Sfakianos, G.; Moschovi, M.; Prodromou, N. Expression of heat shock proteins in medulloblastoma. J. Neurosurg. Pediatr. 2013, 12, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Zagouri, F.; Sergentanis, T.N.; Nonni, A.; Papadimitriou, C.A.; Michalopoulos, N.V.; Domeyer, P.; Theodoropoulos, G.; Lazaris, A.; Patsouris, E.; Zogafos, E.; et al. Hsp90 in the continuum of breast ductal carcinogenesis: Evaluation in precursors, preinvasive and ductal carcinoma lesions. BMC Cancer 2010. [Google Scholar] [CrossRef] [PubMed]
- Nagaraju, G.P.; Long, T.E.; Park, W.; Landry, J.C.; Taliaferro-Smith, L.; Farris, A.B.; Diaz, R.; El Rayes, B.F. Heat shock protein 90 promotes epithelial to mesenchymal transition, invasion, and migration in colorectal cancer. Mol. Carcinog. 2014. [Google Scholar] [CrossRef] [PubMed]
- Annamalai, B.; Liu, X.; Gopal, U.; Isaacs, J.S. Hsp90 is an essential regulator of EphA2 receptor stability and signaling: Implications for cancer cell migration and metastasis. Mol. Cancer Res. 2009, 7, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Kawabe, M.; Mandic, M.; Taylor, J.L.; Vasquez, C.A.; Wesa, A.K.; Neckers, L.M.; Storkus, W.J. Heat shock protein 90 inhibitor 17-dimethylaminoethylamino-17-demethoxygeldanamycin enhances EphA2+ tumor cell recognition by specific CD8+ T cells. Cancer Res. 2009, 69, 6995–7003. [Google Scholar] [CrossRef] [PubMed]
- Bisht, K.S.; Bradbury, C.M.; Mattson, D.; Kaushal, A.; Sowers, A.; Markovina, S.; Ortiz, K.L.; Sieck, L.K.; Isaacs, J.S.; Brechbiel, M.W.; et al. Geldanamycin and 17-allylamino-17-demethoxygeldanamycin potentiate the in vitro and in vivo radiation response of cervical tumor cells via the heat shock protein 90-mediated intracellular signaling and cytotoxicity. Cancer Res. 2003, 63, 8984–8995. [Google Scholar] [PubMed]
- Bull, E.E.; Dote, H.; Brady, K.J.; Burgan, W.E.; Carter, D.J.; Cerra, M.A.; Oswald, K.A.; Hollingshead, M.G.; Camphausen, K.; Tofilon, P.J. Enhanced tumor cell radiosensitivity and abrogation of G2 and S phase arrest by the Hsp90 inhibitor 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin. Clin. Cancer Res. 2004, 10, 8077–8084. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, Y.; Suzuki, S.; Archibald, J.M.; Keeling, P.J.; Ishida, K. Overexpression of molecular chaperone genes in nucleomorph genomes. Mol. Biol. Evol. 2014, 31, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Koll, T.T.; Feis, S.S.; Wright, M.H.; Teniola, M.M.; Richardson, M.M.; Robles, A.I.; Bradsher, J.; Capala, J.; Varticovski, L. HSP90 inhibitor, DMAG, synergizes with radiation of lung cancer cells by interfering with base excision and ATM-mediated DNA repair. Mol. Cancer Ther. 2008, 7, 1985–1992. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, M.; Yu, D.; Hirayama, R.; Ninomiya, Y.; Sekine, E.; Kubota, N.; Ando, K.; Okayasu, R. Inhibition of homologous recombination repair in irradiated tumor cells pretreated with Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Biochem. Biophys. Res. Commun. 2006, 351, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.S.; Burgan, W.; Oswald, K.A.; Camphausen, K.; Tofilon, P.J. Enhanced cell killing induced by the combination of radiation and the heat shock protein 90 inhibitor 17-allylamino-17- demethoxygeldanamycin: A multitarget approach to radiosensitization. Clin. Cancer Res. 2003, 9, 3749–3755. [Google Scholar] [PubMed]
- Workman, P.; Burrows, F.; Neckers, L.; Rosen, N. Drugging the cancer chaperone HSP90: Combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann. NY Acad. Sci. 2007, 1113, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Dote, H.; Burgan, W.E.; Camphausen, K.; Tofilon, P.J. Inhibition of hsp90 compromises the DNA damage response to radiation. Cancer Res. 2006, 66, 9211–9220. [Google Scholar] [CrossRef] [PubMed]
- Han, F.F.; Li, L.; Shang, B.Y.; Shao, R.G.; Zhen, Y.S. Hsp90 inhibitor geldanamycin enhances the antitumor efficacy of enediyne lidamycin in association with reduced DNA damage repair. Asian Pac. J. Cancer Prev. 2014, 15, 7043–7048. [Google Scholar] [CrossRef] [PubMed]
- Ri, M.; Iida, S.; Nakashima, T.; Miyazaki, H.; Mori, F.; Ito, A.; Inagaki, A.; Kusumoto, S.; Ishida, T.; Komatsu, H.; et al. Bortezomib-resistant myeloma cell lines: a Role for mutated PSMB5 in preventing the accumulation of unfolded proteins and fatal ER stress. Leukemia 2010, 24, 1506–1512. [Google Scholar] [CrossRef] [PubMed]
- Robles, A.I.; Wright, M.H.; Gandhi, B.; Feis, S.S.; Hanigan, C.L.; Wiestner, A.; Varticovski, L. Schedule-dependent synergy between the heat shock protein 90 inhibitor 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin and doxorubicin restores apoptosis to p53-mutant lymphoma cell lines. Clin. Cancer Res. 2006, 12, 6547–6556. [Google Scholar] [CrossRef] [PubMed]
- Dehner, A.; Furrer, J.; Richter, K.; Schuster, I.; Buchner, J.; Kessler, H. NMR chemical shift perturbation study of the N-terminal domain of Hsp90 upon binding of ADP, AMP-PNP, geldanamycin, and radicicol. Chembiochem 2003, 4, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Roe, S.M.; Prodromou, C.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J. Med. Chem. 1999, 42, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.H. Small Molecule Inhibitors to Disrupt Protein-protein Interactions of Heat Shock Protein 90 Chaperone Machinery. J. Cancer Prev. 2015, 20, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Stebbins, C.E.; Russo, A.A.; Schneider, C.; Rosen, N.; Hartl, F.U.; Pavletich, N.P. Crystal structure of an Hsp90-geldanamycin complex: Targeting of a protein chaperone by an antitumor agent. Cell 1997, 89, 239–250. [Google Scholar] [CrossRef]
- Jackson, S.E. Hsp90: Structure and function. Top. Curr. Chem. 2013, 328, 155–240. [Google Scholar] [PubMed]
- Supko, J.G.; Hickman, R.L.; Grever, M.R.; Malspeis, L. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother. Pharmacol. 1995, 36, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Banerji, U.; O’Donnell, A.; Scurr, M.; Pacey, S.; Stapleton, S.; Asad, Y.; Simmons, L.; Maloney, A.; Raynaud, F.; Campbell, M.; et al. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. J. Clin. Oncol. 2005, 23, 4152–4161. [Google Scholar] [CrossRef] [PubMed]
- Ronnen, E.A.; Kondagunta, G.V.; Ishill, N.; Sweeney, S.M.; Deluca, J.K.; Schwartz, L.; Bacik, J.; Motzer, R.J. A phase II trial of 17-(Allylamino)-17-demethoxygeldanamycin in patients with papillary and clear cell renal cell carcinoma. Investig. New Drugs 2006, 24, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Smith, V.; Sausville, E.A.; Camalier, R.F.; Fiebig, H.H.; Burger, A.M. Comparison of 17-dimethylaminoethylamino-17-demethoxy-geldanamycin (17DMAG) and 17-allylamino-17-demethoxygeldanamycin (17AAG) in vitro: Effects on Hsp90 and client proteins in melanoma models. Cancer Chemother. Pharmacol. 2005, 56, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Heath, E.I.; Gaskins, M.; Pitot, H.C.; Pili, R.; Tan, W.; Marschke, R.; Liu, G.; Hillman, D.; Sarkar, F.; Sheng, S.; et al. A phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with hormone-refractory metastatic prostate cancer. Clin. Prostate Cancer 2005, 4, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Badros, A.Z.; Jagannath, S.; Tarantolo, S.; Wolf, J.L.; Albitar, M.; Berman, D.; Messina, M.; Anderson, K.C. Tanespimycin with bortezomib: Activity in relapsed/refractory patients with multiple myeloma. Br. J. Haematol. 2010, 150, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Pacey, S.; Gore, M.; Chao, D.; Banerji, U.; Larkin, J.; Sarker, S.; Owen, K.; Asad, Y.; Raynaud, F.; Walton, M.; Judson, I.; Workman, P.; Eisen, T. A Phase II trial of 17-allylamino, 17-demethoxygeldanamycin (17-AAG, tanespimycin) in patients with metastatic melanoma. Investig. New Drugs 2012, 30, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Oki, Y.; Copeland, A.; Romaguera, J.; Fayad, L.; Fanale, M.; Faria, S.C.; Medeiros, L.J.; Ivy, P.; Younes, A. Clinical experience with the heat shock protein-90 inhibitor, tanespimycin, in patients with relapsed lymphoma. Leuk. Lymphoma 2012, 53, 990–992. [Google Scholar] [CrossRef] [PubMed]
- Kitson, R.R.; Chang, C.H.; Xiong, R.; Williams, H.E.; Davis, A.L.; Lewis, W.; Dehn, D.L.; Siegel, D.; Roe, S.M.; Prodromou, C.; et al. Synthesis of 19-substituted geldanamycins with altered conformations and their binding to heat shock protein Hsp90. Nat. Chem. 2013, 5, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zheng, Z.; Zhao, Y.; Zhang, T.; Gu, X.; Yang, W. The heat shock protein 90 inhibitor 17-AAG suppresses growth and induces apoptosis in human cholangiocarcinoma cells. Clin. Exp. Med. 2013, 13, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Hollingshead, M.; Alley, M.; Burger, A.M.; Borgel, S.; Pacula-Cox, C.; Fiebig, H.H.; Sausville, E.A. In vivo antitumor efficacy of 17-DMAG (17-dimethylaminoethylamino-17-demethoxygeldanamycin hydrochloride), a water-soluble geldanamycin derivative. Cancer Chemother. Pharmacol. 2005, 56, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Onuoha, S.C.; Mukund, S.R.; Coulstock, E.T.; Sengerova, B.; Shaw, J.; McLaughlin, S.H.; Jackson, S.E. Mechanistic studies on Hsp90 inhibition by ansamycin derivatives. J. Mol. Biol. 2007, 372, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Pacey, S.; Wilson, R.H.; Walton, M.; Eatock, M.M.; Hardcastle, A.; Zetterlund, A.; Arkenau, H.T.; Moreno-Farre, J.; Banerji, U.; Roels, B.; et al. A phase I study of the heat shock protein 90 inhibitor alvespimycin (17-DMAG) given intravenously to patients with advanced solid tumors. Clin. Cancer Res. 2011, 17, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Zatorska, D.; Kim, J.; Aguirre, J.; Llauger, L.; She, Y.; Wu, N.; Immormino, R.M.; Gewirth, D.T.; Chiosis, G. Identification of potent water soluble purine-scaffold inhibitors of the heat shock protein 90. J. Med. Chem. 2006, 49, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Usmani, S.Z.; Bona, R.D.; Chiosis, G.; Li, Z. The anti-myeloma activity of a novel purine scaffold HSP90 inhibitor PU-H71 is via inhibition of both HSP90A and HSP90B1. J. Hematol. Oncol. 2010, 3. [Google Scholar] [CrossRef] [PubMed]
- Ambati, S.R.; Lopes, E.C.; Kosugi, K.; Mony, U.; Zehir, A.; Shah, S.K.; Taldone, T.; Moreira, A.L.; Meyers, P.A.; Chiosis, G.; et al. Pre-clinical efficacy of PU-H71, a novel HSP90 inhibitor, alone and in combination with bortezomib in Ewing sarcoma. Mol. Oncol. 2014, 8, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Brough, P.A.; Aherne, W.; Barril, X.; Borgognoni, J.; Boxall, K.; Cansfield, J.E.; Cheung, K.M.; Collins, I.; Davies, N.G.; Drysdale, M.J.; et al. 4,5-diarylisoxazole Hsp90 chaperone inhibitors: Potential therapeutic agents for the treatment of cancer. J. Med. Chem. 2008, 51, 196–218. [Google Scholar] [CrossRef] [PubMed]
- Eccles, S.A.; Massey, A.; Raynaud, F.I.; Sharp, S.Y.; Box, G.; Valenti, M.; Patterson, L.; de Haven Brandon, A.; Gowan, S.; Boxall, F.; et al. NVP-AUY922: A novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 2008, 68, 2850–2860. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Tsukuda, K.; Toyooka, S.; Ando, M.; Takaoka, M.; Soh, J.; Asano, H.; Maki, Y.; Muraoka, T.; Tanaka, N.; et al. Strong anti-tumor effect of NVP-AUY922, a novel Hsp90 inhibitor, on non-small cell lung cancer. Lung Cancer 2012, 76, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Ying, W.; Du, Z.; Sun, L.; Foley, K.P.; Proia, D.A.; Blackman, R.K.; Zhou, D.; Inoue, T.; Tatsuta, N.; Sang, J.; et al. Ganetespib, a unique triazolone-containing Hsp90 inhibitor, exhibits potent antitumor activity and a superior safety profile for cancer therapy. Mol. Cancer Ther. 2012, 11, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Casal, R.; Bhattacharya, C.; Epperly, M.W.; Basse, P.H.; Wang, H.; Wang, X.; Proia, D.A.; Greenberger, J.S.; Socinski, M.A.; Levina, V. The HSP90 Inhibitor Ganetespib Radiosensitizes Human Lung Adenocarcinoma Cells. Cancers 2015, 7, 876–907. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Smith, D.L.; Sequeira, M.; Sang, J.; Bates, R.C.; Proia, D.A. The HSP90 inhibitor ganetespib has chemosensitizer and radiosensitizer activity in colorectal cancer. Investig. New Drugs. 2014, 32, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Proia, D.A.; Bates, R.C. Ganetespib and HSP90: Translating preclinical hypotheses into clinical promise. Cancer. Res. 2014, 74, 1294–1300. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera: A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Shiau, A.K.; Harris, S.F.; Southworth, D.R.; Agard, D.A. Structural analysis of E. coli hsp90 reveals dramatic nucleotide-dependent conformational rearrangements. Cell 2006, 127, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.M.; Roe, S.M.; Vaughan, C.K.; Meyer, P.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 2006, 440, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Bridge, G.; Rashid, S.; Martin, S.A. DNA mismatch repair and oxidative DNA damage: Implications for cancer biology and treatment. Cancers 2014, 6, 1597–1614. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Voronova, N.V.; Chistiakov, P.A. Genetic variations in DNA repair genes, radiosensitivity to cancer and susceptibility to acute tissue reactions in radiotherapy-treated cancer patients. Acta Oncol. 2008, 47, 809–824. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Dezwaan, D.C.; Freeman, B.C. HSP90: The rosetta stone for cellular protein dynamics? Cell Cycle 2008, 7, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Quanz, M.; Herbette, A.; Sayarath, M.; de Koning, L.; Dubois, T.; Sun, J.S.; Dutreix, M. Heat shock protein 90α (Hsp90α) is phosphorylated in response to DNA damage and accumulates in repair foci. J. Biol. Chem. 2012, 287, 8803–8815. [Google Scholar] [CrossRef] [PubMed]
- Solier, S.; Kohn, K.W.; Scroggins, B.; Xu, W.; Trepel, J.; Neckers, L.; Pommier, Y. Heat shock protein 90alpha (HSP90α), a substrate and chaperone of DNA-PK necessary for the apoptotic response. Proc. Natl. Acad. Sci. USA 2012, 109, 12866–12872. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Horikoshi, N.; Singh, M.; Gupta, A.; Misra, H.S.; Albuquerque, K.; Hunt, C.R.; Pandita, T.K. Chromatin modifications and the DNA damage response to ionizing radiation. Front. Oncol. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Bassing, C.H.; Suh, H.; Ferguson, D.O.; Chua, K.F.; Manis, J.; Eckersdorff, M.; Gleason, M.; Bronson, R.; Lee, C.; Alt, F.W. Histone H2AX: A dosage-dependent suppressor of oncogenic translocations and tumors. Cell 2003, 114, 359–370. [Google Scholar] [CrossRef]
- Scully, R.; Xie, A. Double strand break repair functions of histone H2AX. Mutat. Res. 2013, 750, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Kanaar, R.; Hoeijmakers, J.H.; van Gent, D.C. Molecular mechanisms of DNA double strand break repair. Trends Cell Biol. 1998, 8, 483–489. [Google Scholar] [CrossRef]
- Lieber, M.R.; Ma, Y.; Pannicke, U.; Schwarz, K. Mechanism and regulation of human non-homologous DNA end-joining. Nat. Rev. Mol. Cell Biol. 2003, 4, 712–720. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, M.; Jeggo, P.A. The role of double-strand break repair—Insights from human genetics. Nat. Rev. Genet. 2006, 7, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Van Gent, D.C.; Hoeijmakers, J.H.; Kanaar, R. Chromosomal stability and the DNA double-stranded break connection. Nat. Rev. Genet. 2001, 2, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, C.; Soutoglou, E. Double strand break (DSB) repair in heterochromatin and heterochromatin proteins in DSB repair. DNA Repair 2014, 19, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Pierce, A.J.; Jasin, M. NHEJ deficiency and disease. Mol. Cell 2001, 8, 1160–1161. [Google Scholar] [CrossRef]
- Rothkamm, K.; Kruger, I.; Thompson, L.H.; Lobrich, M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell. Biol. 2003, 23, 5706–5715. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Jeggo, P.A. The repair and signaling responses to DNA double-strand breaks. Adv. Genet. 2013, 82, 1–45. [Google Scholar] [PubMed]
- Dynan, W.S.; Yoo, S. Interaction of Ku protein and DNA-dependent protein kinase catalytic subunit with nucleic acids. Nucleic Acids Res. 1998, 26, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Pannicke, U.; Schwarz, K.; Lieber, M.R. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell 2002, 108, 781–794. [Google Scholar] [CrossRef]
- Gupta, A.; Hunt, C.R.; Chakraborty, S.; Pandita, R.K.; Yordy, J.; Ramnarain, D.B.; Horikoshi, N.; Pandita, T.K. Role of 53BP1 in the regulation of DNA double-strand break repair pathway choice. Radiat. Res. 2014, 181, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Di Masi, A.; Gullotta, F.; Cappadonna, V.; Leboffe, L.; Ascenzi, P. Cancer predisposing mutations in BRCT domains. IUBMB Life 2011, 63, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Arlander, S.J.; Eapen, A.K.; Vroman, B.T.; McDonald, R.J.; Toft, D.O.; Karnitz, L.M. Hsp90 inhibition depletes Chk1 and sensitizes tumor cells to replication stress. J. Biol. Chem. 2003, 278, 52572–52577. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Inanc, B.; Schamus, S.; Wang, X.H.; Wei, L.; Brown, A.R.; Svilar, D.; Sugrue, K.F.; Goellner, E.M.; Zeng, X.; et al. HSP90 regulates DNA repair via the interaction between XRCC1 and DNA polymerase beta. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Furuta, T.; Takemura, H.; Liao, Z.Y.; Aune, G.J.; Redon, C.; Sedelnikova, O.A.; Pilch, D.R.; Rogakou, E.P.; Celeste, A.; Chen, H.T.; et al. Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian DNA topoisomerase I cleavage complexes. J. Biol. Chem. 2003, 278, 20303–20312. [Google Scholar] [CrossRef] [PubMed]
- Schnaider, T.; Oikarinen, J.; Ishiwatari-Hayasaka, H.; Yahara, I.; Csermely, P. Interactions of Hsp90 with histones and related peptides. Life Sci. 1999, 65, 2417–2426. [Google Scholar] [CrossRef]
- Smogorzewska, A.; Matsuoka, S.; Vinciguerra, P.; McDonald, E.R., III; Hurov, K.E.; Luo, J.; Ballif, B.A.; Gygi, S.P.; Hofmann, K.; D’Andrea, A.D.; et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 2007, 129, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Stecklein, S.R.; Kumaraswamy, E.; Behbod, F.; Wang, W.; Chaguturu, V.; Harlan-Williams, L.M.; Jensen, R.A. BRCA1 and HSP90 cooperate in homologous and non-homologous DNA double-strand-break repair and G2/M checkpoint activation. Proc. Natl. Acad. Sci. USA 2012, 109, 13650–13655. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, X.; Wang, P.; Yu, X.; Essers, J.; Chen, D.; Kanaar, R.; Takeda, S.; Wang, Y. Characteristics of DNA-binding proteins determine the biological sensitivity to high-linear energy transfer radiation. Nucleic Acids Res. 2010, 38, 3245–3251. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Mimnaugh, E.G.; de Costa, B.; Myers, C.E.; Neckers, L.M. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: Essential role for stress proteins in oncogenic transformation. Proc. Natl. Acad. Sci. USA 1994, 91, 8324–8328. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, T.; Schwartz, S.J.; Sun, D. New developments in Hsp90 inhibitors as anti-cancer therapeutics: Mechanisms, clinical perspective and more potential. Drug Resist. Updat. 2009, 12, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Pacey, S.; Banerji, U.; Judson, I.; Workman, P. Hsp90 inhibitors in the clinic. Handb. Exp. Pharmacol. 2006, 331–358. [Google Scholar]
- Chiosis, G. Targeting chaperones in transformed systems—A focus on Hsp90 and cancer. Expert. Opin. Ther. Targets 2006, 10, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Solit, D.B.; Chiosis, G. Development and application of Hsp90 inhibitors. Drug Discov. Today 2008, 13, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.M.; Matthews, T.P.; James, K.; Rowlands, M.G.; Boxall, K.J.; Sharp, S.Y.; Maloney, A.; Roe, S.M.; Prodromou, C.; Pearl, L.H.; et al. The identification, synthesis, protein crystal structure and in vitro biochemical evaluation of a new 3,4-diarylpyrazole class of Hsp90 inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 3338–3343. [Google Scholar] [CrossRef] [PubMed]
- McDonald, E.; Jones, K.; Brough, P.A.; Drysdale, M.J.; Workman, P. Discovery and development of pyrazole-scaffold Hsp90 inhibitors. Curr. Top. Med. Chem. 2006, 6, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L. Evolution and function of diverse Hsp90 homologs and cochaperone proteins. Biochim. Biophys. Acta 2012, 1823, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Ernst, A.; Anders, H.; Kapfhammer, H.; Orth, M.; Hennel, R.; Seidl, K.; Winssinger, N.; Belka, C.; Unkel, S.; Lauber, K. HSP90 inhibition as a means of radiosensitizing resistant, aggressive soft tissue sarcomas. Cancer Lett. 2015, 365, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Falsone, S.F.; Gesslbauer, B.; Tirk, F.; Piccinini, A.M.; Kungl, A.J. A proteomic snapshot of the human heat shock protein 90 interactome. FEBS Lett. 2005, 579, 6350–6354. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.; McLaughlin, M.; Bhide, S.A.; Eccles, S.A.; Workman, P.; Nutting, C.M.; Huddart, R.A.; Harrington, K.J. The HSP90 inhibitor NVP-AUY922 radiosensitizes by abrogation of homologous recombination resulting in mitotic entry with unresolved DNA damage. PLoS ONE 2012, 7, e35436. [Google Scholar] [CrossRef] [PubMed]
- Parameswaran, B.; Chiang, H.C.; Lu, Y.; Coates, J.; Deng, C.X.; Baer, R.; Lin, H.K.; Li, R.; Paull, T.T.; Hu, Y. Damage-induced BRCA1 phosphorylation by Chk2 contributes to the timing of end resection. Cell Cycle 2015, 14, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D.; Wang, Y.; Qin, J.; Elledge, S.J. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science 1999, 286, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Yarden, R.I.; Pardo-Reoyo, S.; Sgagias, M.; Cowan, K.H.; Brody, L.C. BRCA1 regulates the G2/M checkpoint by activating Chk1 kinase upon DNA damage. Nat. Genet. 2002, 30, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, A.; Ear, U.S.; Koller, B.H.; Weichselbaum, R.R.; Bishop, D.K. The breast cancer susceptibility gene BRCA1 is required for subnuclear assembly of Rad51 and survival following treatment with the DNA cross-linking agent cisplatin. J. Biol. Chem. 2000, 275, 23899–23903. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Nievera, C.J.; Lee, A.Y.; Wu, X. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J. Biol. Chem. 2008, 283, 7713–7720. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, A.D.; Grompe, M. The Fanconi anaemia/BRCA pathway. Nat .Rev. Cancer 2003, 3, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Chen, C.F.; Li, S.; Chen, Y.; Wang, C.C.; Xiao, J.; Chen, P.L.; Sharp, Z.D.; Lee, W.H. Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. Science 1999, 285, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Scata, K.A.; El Deiry, W.S. p53, BRCA1 and breast Cancer chemoresistance. Adv. Exp. Med. Biol. 2007, 608, 70–86. [Google Scholar] [PubMed]
- Davies, A.A.; Masson, J.Y.; McIlwraith, M.J.; Stasiak, A.Z.; Stasiak, A.; Venkitaraman, A.R.; West, S.C. Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol. Cell 2001, 7, 273–282. [Google Scholar] [CrossRef]
- Segawa, T.; Fujii, Y.; Tanaka, A.; Bando, S.; Okayasu, R.; Ohnishi, K.; Kubota, N. Radiosensitization of human lung cancer cells by the novel purine-scaffold Hsp90 inhibitor, PU-H71. Int. J. Mol. Med. 2014, 33, 559–564. [Google Scholar] [PubMed]
- Dungey, F.A.; Caldecott, K.W.; Chalmers, A.J. Enhanced radiosensitization of human glioma cells by combining inhibition of poly(ADP-ribose) polymerase with inhibition of heat shock protein 90. Mol. Cancer Ther. 2009, 8, 2243–2254. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Weigel, B.; Kersey, J. Synergism between etoposide and 17-AAG in leukemia cells: Critical roles for Hsp90, FLT3, topoisomerase II, Chk1, and Rad51. Clin. Cancer Res. 2007, 13, 1591–1600. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [PubMed]
- Dai, Y.; Grant, S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin. Cancer Res. 2010, 16, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Oakley, G.G.; Patrick, S.M. Replication protein A: Directing traffic at the intersection of replication and repair. Fron. Biosci. 2010, 15, 883–900. [Google Scholar] [CrossRef]
- Gonzalez Besteiro, M.A.; Gottifredi, V. The fork and the kinase: A DNA replication tale from a CHK1 perspective. Mutat. Res. Rev. Mutat. Res. 2015, 763, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Roos-Mattjus, P.; Hopkins, K.M.; Oestreich, A.J.; Vroman, B.T.; Johnson, K.L.; Naylor, S.; Lieberman, H.B.; Karnitz, L.M. Phosphorylation of human Rad9 is required for genotoxin-activated checkpoint signaling. J. Biol. Chem. 2003, 278, 24428–24437. [Google Scholar] [CrossRef] [PubMed]
- Zachos, G.; Rainey, M.D.; Gillespie, D.A. Chk1-deficient tumour cells are viable but exhibit multiple checkpoint and survival defects. EMBO J. 2003, 22, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Dittmann, K.; Mayer, C.; Fehrenbacher, B.; Schaller, M.; Raju, U.; Milas, L.; Chen, D.J.; Kehlbach, R.; Rodemann, H.P. Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of DNA-dependent protein kinase. J. Biol. Chem. 2005, 280, 31182–31189. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, R.; Solary, E.; O’Connor, P.; Kohn, K.W.; Pommier, Y. Induction of a common pathway of apoptosis by staurosporine. Exp. Cell Res. 1994, 211, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Strasser, A.; Jost, P.J.; Nagata, S. The many roles of FAS receptor signaling in the immune system. Immunity 2009, 30, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Solier, S.; Pommier, Y. The apoptotic ring: A novel entity with phosphorylated histones H2AX and H2B and activated DNA damage response kinases. Cell Cycle 2009, 8, 1853–1859. [Google Scholar] [CrossRef] [PubMed]
- Solier, S.; Sordet, O.; Kohn, K.W.; Pommier, Y. Death receptor-induced activation of the Chk2- and histone H2AX-associated DNA damage response pathways. Mol. Cell. Biol. 2009, 29, 68–82. [Google Scholar] [CrossRef] [PubMed]
- Vasilevskaya, I.A.; O’Dwyer, P.J. 17-Allylamino-17-demethoxygeldanamycin overcomes TRAIL resistance in colon cancer cell lines. Biochem. Pharmacol. 2005, 70, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Solier, S.; Pommier, Y. MDC1 cleavage by caspase-3: A novel mechanism for inactivating the DNA damage response during apoptosis. Cancer Res. 2011, 71, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Yang, Y.G.; Pierce, A.J.; Taniguchi, T.; Digweed, M.; D’Andrea, A.D.; Wang, Z.Q.; Jasin, M. Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proc. Natl. Acad. Sci. USA 2005, 102, 1110–1115. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Ishiai, M.; Matsushita, N.; Arakawa, H.; Lamerdin, J.E.; Buerstedde, J.M.; Tanimoto, M.; Harada, M.; Thompson, L.H.; Takata, M. Fanconi anemia FANCG protein in mitigating radiation- and enzyme-induced DNA double-strand breaks by homologous recombination in vertebrate cells. Mol. Cell. Biol. 2003, 23, 5421–5430. [Google Scholar] [CrossRef] [PubMed]
- Oda, T.; Hayano, T.; Miyaso, H.; Takahashi, N.; Yamashita, T. Hsp90 regulates the Fanconi anemia DNA damage response pathway. Blood 2007, 109, 5016–5026. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Oda, T.; Sekimoto, T. Hsp90 and the Fanconi anemia pathway: A molecular link between protein quality control and the DNA damage response. Cell Cycle 2007, 6, 2232–2235. [Google Scholar] [CrossRef] [PubMed]
- Gospodinov, A.; Herceg, Z. Chromatin structure in double strand break repair. DNA Repair 2013, 12, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Jeggo, P.A.; Downs, J.A. Roles of chromatin remodellers in DNA double strand break repair. Exp. Cell Res. 2014, 329, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Csordas, A. On the biological role of histone acetylation. Biochem. J. 1990, 265, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.S.; Rimmer, J.M.; Green, B.N.; Finch, J.T.; Thomas, J.O. Histone-DNA interactions and their modulation by phosphorylation of -Ser-Pro-X-Lys/Arg- motifs. EMBO J. 1991, 10, 1939–1948. [Google Scholar] [PubMed]
- Khadake, J.R.; Rao, M.R. Condensation of DNA and chromatin by an SPKK-containing octapeptide repeat motif present in the C-terminus of histone H1. Biochemistry 1997, 36, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Tarkka, T.; Oikarinen, J.; Grundstrom, T. Nucleotide and calcium-induced conformational changes in histone H1. FEBS Lett. 1997, 406, 56–60. [Google Scholar] [CrossRef]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Mo, C.; Dai, Y.; Kang, N.; Cui, L.; He, W. Ectopic expression of human MutS homologue 2 on renal carcinoma cells is induced by oxidative stress with interleukin-18 promotion via p38 mitogen-activated protein kinase (MAPK) and c-Jun N-terminal kinase (JNK) signaling pathways. J. Biol. Chem. 2012, 287, 19242–19254. [Google Scholar] [CrossRef] [PubMed]
- Tung, H.Y.; Plunkett, B.; Huang, S.K.; Zhou, Y. Murine mast cells secrete and respond to interleukin-33. J. Interferon Cytokine Res. 2014, 34, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J.; Harris, A.L.; Aherne, G.W. Mechanism of cell death following thymidylate synthase inhibition: 2′-deoxyuridine-5′-triphosphate accumulation, DNA damage, and growth inhibition following exposure to CB3717 and dipyridamole. Cancer Res. 1991, 51, 2346–2352. [Google Scholar] [PubMed]
- Van der Wilt, C.L.; Kuiper, C.M.; Peters, G.J. Combination studies of antifolates with 5-fluorouracil in colon cancer cell lines. Oncol. Res. 1999, 11, 383–391. [Google Scholar] [PubMed]
- White, I.N. Tamoxifen: Is it safe? Comparison of activation and detoxication mechanisms in rodents and in humans. Curr. Drug Metab. 2003, 4, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Olivo-Marston, S.E.; Mechanic, L.E.; Mollerup, S.; Bowman, E.D.; Remaley, A.T.; Forman, M.R.; Skaug, V.; Zheng, Y.L.; Haugen, A.; Harris, C.C. Serum estrogen and tumor-positive estrogen receptor-alpha are strong prognostic classifiers of non-small-cell lung cancer survival in both men and women. Carcinogenesis 2010, 31, 1778–1786. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.C.; Chiu, H.C.; Syu, J.J.; Jian, Y.J.; Chen, C.Y.; Jian, Y.T.; Huang, Y.J.; Wo, T.Y.; Lin, Y.W. Tamoxifen enhances erlotinib-induced cytotoxicity through down-regulating AKT-mediated thymidine phosphorylase expression in human non-small-cell lung cancer cells. Biochem. Pharmacol. 2014, 88, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.C.; Chiu, H.C.; Syu, J.J.; Chen, C.Y.; Jian, Y.T.; Huang, Y.J.; Wo, T.Y.; Jian, Y.J.; Chang, P.Y.; Wang, T.J.; Lin, Y.W. Down-regulation of MSH2 expression by Hsp90 inhibition enhances cytotoxicity affected by tamoxifen in human lung cancer cells. Biochem. Biophys. Res. Commun. 2015, 456, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C.; Lehmann, A.R.; Fuchs, R.P. Trading places: How do DNA polymerases switch during translesion DNA synthesis? Mol. Cell 2005, 18, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, A.R.; Niimi, A.; Ogi, T.; Brown, S.; Sabbioneda, S.; Wing, J.F.; Kannouche, P.L.; Green, C.M. Translesion synthesis: Y-family polymerases and the polymerase switch. DNA Repair 2007, 6, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Woodgate, R. What a difference a decade makes: Insights into translesion DNA synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 15591–15598. [Google Scholar] [CrossRef] [PubMed]
- Masutani, C.; Kusumoto, R.; Iwai, S.; Hanaoka, F. Mechanisms of accurate translesion synthesis by human DNA polymerase eta. EMBO J. 2000, 19, 3100–3109. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, S.D.; Kokoska, R.J.; Kunkel, T.A. Efficiency, fidelity and enzymatic switching during translesion DNA synthesis. Cell Cycle 2004, 3, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Kannouche, P.L.; Wing, J.; Lehmann, A.R. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: A possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell 2004, 14, 491–500. [Google Scholar] [CrossRef]
- Watanabe, K.; Tateishi, S.; Kawasuji, M.; Tsurimoto, T.; Inoue, H.; Yamaizumi, M. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004, 23, 3886–3896. [Google Scholar] [CrossRef] [PubMed]
- Sekimoto, T.; Oda, T.; Pozo, F.M.; Murakumo, Y.; Masutani, C.; Hanaoka, F.; Yamashita, T. The molecular chaperone Hsp90 regulates accumulation of DNA polymerase eta at replication stalling sites in UV-irradiated cells. Mol. Cell 2010, 37, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Heuvelman, D.M.; Carroll, J.A.; Dufield, D.R.; Masferrer, J.L. Geldanamycin-induced PCNA degradation in isolated Hsp90 complex from cancer cells. Cancer Investig. 2010, 28, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T.; Prigent, C.; Barnes, D.E.; Lehmann, A.R.; Satoh, M.S.; Roberts, E.; Nash, R.A.; Robins, P.; Daly, G. DNA joining in mammalian cells. Cold Spring Harb. Symp. Quant. Biol. 1993, 58, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Wilson, D.M., III. Overview of base excision repair biochemistry. Curr. Mol. Pharmacol. 2012, 5, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Svilar, D.; Goellner, E.M.; Almeida, K.H.; Sobol, R.W. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid. Redox Signal. 2011, 14, 2491–2507. [Google Scholar] [CrossRef] [PubMed]
- Braithwaite, E.K.; Kedar, P.S.; Stumpo, D.J.; Bertocci, B.; Freedman, J.H.; Samson, L.D.; Wilson, S.H. DNA polymerases beta and lambda mediate overlapping and independent roles in base excision repair in mouse embryonic fibroblasts. PLoS ONE 2010, 5, e12229. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. XRCC1 and DNA strand break repair. DNA Repair 2003, 2, 955–969. [Google Scholar] [CrossRef]
- Shim, H.J.; Yun, J.Y.; Hwang, J.E.; Bae, W.K.; Cho, S.H.; Lee, J.H.; Kim, H.N.; Shin, M.H.; Kweon, S.S.; Lee, J.H.; et al. BRCA1 and XRCC1 polymorphisms associated with survival in advanced gastric cancer treated with taxane and cisplatin. Cancer Sci. 2010, 101, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [PubMed]
- Brem, R.; Hall, J. XRCC1 is required for DNA single-strand break repair in human cells. Nucleic Acids Res. 2005, 33, 2512–2520. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Dittmann, K.; Fehrenbacher, B.; Schaller, M.; Baumann, M.; Rodemann, H.P. PI3K-Akt signaling regulates basal, but MAP-kinase signaling regulates radiation-induced XRCC1 expression in human tumor cells in vitro. DNA Repair 2008, 7, 1746–1756. [Google Scholar] [CrossRef] [PubMed]
- Della-Maria, J.; Zhou, Y.; Tsai, M.S.; Kuhnlein, J.; Carney, J.P.; Paull, T.T.; Tomkinson, A.E. Human Mre11/human Rad50/Nbs1 and DNA ligase IIIalpha/XRCC1 protein complexes act together in an alternative nonhomologous end joining pathway. J. Biol. Chem. 2011, 286, 33845–33853. [Google Scholar] [CrossRef] [PubMed]
- Saribasak, H.; Maul, R.W.; Cao, Z.; McClure, R.L.; Yang, W.; McNeill, D.R.; Wilson, D.M., III; Gearhart, P.J. XRCC1 suppresses somatic hypermutation and promotes alternative nonhomologous end joining in Igh genes. J. Exp. Med. 2011, 208, 2209–2216. [Google Scholar] [CrossRef] [PubMed]
- Tung, C.L.; Jian, Y.J.; Syu, J.J.; Wang, T.J.; Chang, P.Y.; Chen, C.Y.; Jian, Y.T.; Lin, Y.W. Down-regulation of ERK1/2 and AKT-mediated X-ray repair cross-complement group 1 protein (XRCC1) expression by Hsp90 inhibition enhances the gefitinib-induced cytotoxicity in human lung cancer cells. Exp. Cell. Res. 2015, 334, 126–135. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pennisi, R.; Ascenzi, P.; Di Masi, A. Hsp90: A New Player in DNA Repair? Biomolecules 2015, 5, 2589-2618. https://doi.org/10.3390/biom5042589
AMA Style
Pennisi R, Ascenzi P, Di Masi A. Hsp90: A New Player in DNA Repair? Biomolecules. 2015; 5(4):2589-2618. https://doi.org/10.3390/biom5042589
Chicago/Turabian StylePennisi, Rosa, Paolo Ascenzi, and Alessandra Di Masi. 2015. "Hsp90: A New Player in DNA Repair?" Biomolecules 5, no. 4: 2589-2618. https://doi.org/10.3390/biom5042589