





Abstract
SILVA (from Latin silva, forest, http://www.arb-silva.de) is a comprehensive web resource for up to date, quality-controlled databases of aligned ribosomal RNA (rRNA) gene sequences from the Bacteria, Archaea and Eukaryota domains and supplementary online services. The referred database release 111 (July 2012) contains 3 194 778 small subunit and 288 717 large subunit rRNA gene sequences. Since the initial description of the project, substantial new features have been introduced, including advanced quality control procedures, an improved rRNA gene aligner, online tools for probe and primer evaluation and optimized browsing, searching and downloading on the website. Furthermore, the extensively curated SILVA taxonomy and the new non-redundant SILVA datasets provide an ideal reference for high-throughput classification of data from next-generation sequencing approaches.
INTRODUCTION
Sequencing the ribosomal RNA gene (rRNA) is the method of choice for nucleic acid-based detection and identification of microbes, their taxonomic assignment, phylogenetic analysis and the investigation of microbial diversity. Consequently, vast amounts of rRNA gene sequence data—more than 3.5 million sequences (July 2012)—have been accumulated and are publicly available via the International Nucleotide Sequence Database Collaboration (INSDC) databases (1). While the quantity of data further increases the relevance of rRNA genes for marker gene studies for all domains of life, it also creates significant challenges for data management and curation. For optimal utility, the sequences must be extracted and checked for quality, the annotations must be updated and extended to reflect current understanding and finally all the data must be prepared in a coherent, easily accessible manner. These tasks are beyond the scope of the INSDC databases and therefore performed by domain-specific databases. The Ribosomal Database Project (RDP-II) (2,3) and greengenes (4) both cover the domains Archaea and Bacteria for small subunit rRNA gene (SSU) sequences. The SILVA project also includes the Eukaryota, thus covering all three domains of life. Furthermore, SILVA offers databases for both the SSU and the large subunit rRNA gene (LSU).
The SILVA databases are made available as releases, rather than being updated continuously, to enhance the comparability of the studies employing these databases. Each release is numbered according to the EMBL-Bank release from which the sequence data were extracted and is permanently available for download via the SILVA website. Best efforts are made to provide two full releases per year. The database releases are structured into two datasets for each gene: SILVA Parc and SILVA Ref. The Parc datasets comprise the entire SILVA databases for the respective gene, whereas the Ref datasets represent a subset of the Parc comprising only high-quality nearly full-length sequences.
All SILVA datasets contain a rich set of contextual and sequence-associated information. This includes taxonomic classifications from several taxonomy providers, a multiple sequence alignment, type strain information and the latest valid nomenclature. All sequences are quality checked. The corresponding data are made available as ARB files (5) as well as in FASTA and comma-separated value (CSV) formats. Finally, they can be browsed directly via the SILVA website. The combination of SILVA datasets with the ARB software suite provides an advanced workbench for researchers to perform in-depth sequence analysis and phylogenetic reconstructions, as well as manual curation of rRNA gene datasets. The flat-file exports of the SILVA datasets make it easy to integrate SILVA as a source for reference data in next-generation sequencing (NGS) analysis pipelines such as MOTHUR (6), QIIME (7) or MG-RAST (8).
Since its first release in 2007, 16 full releases have been published by the SILVA project. Many improvements to both the release preparation process and the features offered by the project website (http://www.arb-silva.de) have been made. The group of SILVA users has grown to include thousands of researchers worldwide who visit the website regularly to obtain the recent database releases and to employ the SILVA online services in their work. In the first part of this update paper, we describe the most significant changes to data processing within SILVA. The second part outlines the new or improved functions available on the SILVA website.
DATABASES
rRNA gene prediction
The selection of rRNA gene candidate sequences based on annotations in the EMBL-Bank source database is now complemented by hidden Markov model-based rRNA gene prediction. All sequences in EMBL-Bank are scanned for rRNA gene sequences using the models and parameters from the RNAmmer software package (9), HMMER2 (http://hmmer.janelia.org/) and a custom pipeline component. The gene boundaries of the predicted rRNA gene are determined during sequence alignment. Conflicts between EMBL-Bank annotations and predictions are resolved by giving priority to the EMBL-Bank annotation. The source for the SILVA annotation is documented in the field ann_src_slv. As of release 111, the SILVA SSU database contains 53 950 sequences detected solely by the RNAmmer models and 1 537 342 sequences that were both annotated as rRNA and detected by the RNAmmer models. The LSU database contains 17 828 sequences detected solely by RNAmmer models and 17 563 sequences both annotated as rRNA and detected by the RNAmmer models.
Quality control/quality assurance
Sequences with or
for any criterion are rejected.
The ‘Alignment Quality’ of a sequence is determined by three values reported by the SINA alignment software (10): the alignment score, the base pair score and, as of release 111, the alignment identity (for details, see ‘Aligner’ section). Sequences are rejected based on these values to achieve specificity of the SILVA databases, correcting over-prediction by the RNAmmer models as well as removing sequences wrongly annotated as rRNA genes. The alignment quality thresholds for the different datasets can be found in Table 1.
List of alignment quality thresholds used to exclude sequences from the different SILVA datasets
| LSU Parc | LSU Ref | SSU Parc | SSU Ref | |
|---|---|---|---|---|
| Alignment length (bp) | 300 | 1900 | 300 | Bacteria/Eukaryota: 1200 Archaea: 900 |
| Alignment identity (%) | 40 | 60 | 50 | 70 |
| Alignment score (quality) | 30 | 30 | 40 | 50 |
| Base pair score | 30 | 30 | 30 | 30 |
| LSU Parc | LSU Ref | SSU Parc | SSU Ref | |
|---|---|---|---|---|
| Alignment length (bp) | 300 | 1900 | 300 | Bacteria/Eukaryota: 1200 Archaea: 900 |
| Alignment identity (%) | 40 | 60 | 50 | 70 |
| Alignment score (quality) | 30 | 30 | 40 | 50 |
| Base pair score | 30 | 30 | 30 | 30 |
List of alignment quality thresholds used to exclude sequences from the different SILVA datasets
| LSU Parc | LSU Ref | SSU Parc | SSU Ref | |
|---|---|---|---|---|
| Alignment length (bp) | 300 | 1900 | 300 | Bacteria/Eukaryota: 1200 Archaea: 900 |
| Alignment identity (%) | 40 | 60 | 50 | 70 |
| Alignment score (quality) | 30 | 30 | 40 | 50 |
| Base pair score | 30 | 30 | 30 | 30 |
| LSU Parc | LSU Ref | SSU Parc | SSU Ref | |
|---|---|---|---|---|
| Alignment length (bp) | 300 | 1900 | 300 | Bacteria/Eukaryota: 1200 Archaea: 900 |
| Alignment identity (%) | 40 | 60 | 50 | 70 |
| Alignment score (quality) | 30 | 30 | 40 | 50 |
| Base pair score | 30 | 30 | 30 | 30 |
The thresholds upon which sequences are rejected are based on the statistical analyses performed in Schweer (11). They were selected as a conservative balance between rejecting too many valid sequences and keeping too many questionable sequences. A common threshold of 2% was found to be best for the sequence quality metrics.
Prediction of potentially anomalous sequences, such as chimeras, remains unchanged with respect to the original SILVA publication. No filtering is performed using this metric due to the difficulty of clearly differentiating between artefacts of the sequence acquisition process and unusual yet natural evolutionary events. The correct choice—whether and at which threshold potentially anomalous sequences need to be excluded—should be made in light of the specific research question and the experimental setup. We must therefore relegate such filtering to the individual researchers.
SILVA taxonomy
A substantial revision of the classification of all bacterial and archaeal sequences in the Ref datasets was first published with SILVA release 100. Based on the ‘guide trees’, all taxonomic assignments are manually curated and follow the Bergey’s Manual of Systematic Bacteriology (12). Specifically, Archaea, Cyanobacteria, Chloroflexi and Chlorobi are based on volume 1; Proteobacteria on volume 2; Firmicutes on volume 3; Bacteroidetes, Spirochaetes, Tenericutes (Mollicutes), Acidobacteria, Fibrobacteres, Fusobacteria, Dictyoglomi, Gemmatimonadetes, Lentisphaerae, Verrucomicrobia, Chlamydiae and Planctomycetes on volume 4 and finally Actinobacteria on volume 5. Since taxonomy and species are dynamic entities with rapid turnover, name changes and taxonomic outlines are also adapted from List of Prokaryotic Names with Standing in Nomenclature (LPSN) (13).
Although the classification is mainly based on these authoritative resources, deviations from their recommendations do exist: the classification is a phylogenetic tree-based process and differences from the original description and classifications are to be expected. For example, the genus Ahrensia (type species accession: D88524) is classified under family Rhodobacteraceae of Alphaproteobacteria; however, in the SSU Ref guide tree, this genus is grouped together with members of family Phyllobacteriaceae. Normally, such discrepancies are accommodated by introducing polyphyletic groups; however, in this case genus Ahrensia is classified within Phyllobacteriaceae due to high sequence identities (>94%) observed with other members of this family.
The LPSN resource is further used to track down names without standing in nomenclature (not-validly published taxa) and Candidatus taxa. The inclusion of the two latter categories is a unique feature of the SILVA taxonomy. Furthermore, collaborations with domain experts have been established to annotate uncultured clades. A number of examples are the OCS116 clade (14), the SAGMC and SAGME groups (15) and the termite clusters (16).
For an improved and unified taxonomy for Eukaryota based on 18S rRNA gene sequences, the Eukaryotic Taxonomy Working Group (ETWG) has been founded in October 2011. The first version of the new eukaryotic taxonomy was deployed with SILVA release 111. Specifically, the taxonomy of protist lineages has been reconciled with the International Society of Protistologists (ISOP) publication (17). An early draft from the ISOP committee (Adl et al. 2012, manuscript in preparation) was used to further improve protist classification where possible. Higher-level ranks have been revised for higher plants, fungi and animals. By implementing the classification of ISOP, their concept of ‘rankless’ taxa was introduced to the SILVA taxonomy. That is, the position of a taxon in the taxonomic hierarchy does no longer necessarily imply a rank. Although this concept is biologically sound, we recognize the difficulties that this may bring in computational analyses. Therefore, a file containing classification rank mappings is provided with the new eukaryotic taxonomy. These mappings assign reasonable ranks to taxa in order to make the different levels comparable.
Third-party contextual data
Several fields containing additional contextual data have been added to the SILVA databases over the last years. Basic fields include organism name, author, title, publication ID, collection, submission and modification dates as well as latitude/longitude, depth, habitat and taxonomic classifications by various other databases. Tables detailing the fields available in the current release can be found in the Frequently Asked Questions (FAQ) section of the webpage (http://www.arb-silva.de/documentation/background/faqs/).
The ‘strain’ field, carrying the strain data imported from EMBL-Bank, is now extended with information from third-party sources. In addition to the tag ‘(T)’ used by EMBL-Bank to mark a sequence as type strain, the following tags are used by SILVA:
the label ‘e[G]’ is added if an entry is part of the list of genomes offered by the EMBL-Bank,
the label ‘l[T]’ is added if the entry is part of the type strain datasets of ‘The All-Species Living Tree‘project (18,19),
the label ‘s[T]’ is added if an entry is listed as a type strain by the StrainInfo project (20),
the label ‘s[C]’ is added if an entry is a cultured strain according to the StrainInfo project and
the label ‘r[T]’ is added if an entry is listed as a type strain by the RDP-II project.
Furthermore, manually curated habitat descriptors and other contextual information are incorporated where available based on information provided by the megx.net project (21).
Datasets
Ref
The basic criteria for inclusion of sequences in the high-quality full-length Ref datasets have remained unchanged since 2007. Briefly, for SSU Ref archaeal sequences must have at least 900 bases length, bacterial and eukaryotic sequences must have at least 1200 bases length and all sequences must have an alignment score of at least 50. Since release 111, sequences must also have an alignment identity score of at least 70. Furthermore, sequences from large-scale submissions such as made by the mouse wound microbiome project, the human skin microbiome project or the Guerrero Negro hypersaline microbial mat project are removed from the SSU Ref and provided in a separate dataset. Please refer to the SILVA website for information on which projects were removed from each respective database release. Criteria for LSU Ref datasets can be found in Table 1.
SSU Ref NR
For users interested in a representative collection of SSU rRNA gene sequences, the SILVA project offers a non-redundant (NR) version of the SSU Ref dataset. The Ref NR dataset is created by clustering at 99% (up to SILVA 108) and 98% (SILVA 111) sequence identity using UCLUST (22). Of each cluster, only the longest sequence is kept. Sequences from cultivated species including type strains and multiple operons are preserved in all cases to serve as an anchor for taxonomy. The resulting SSU Ref NR dataset is significantly smaller (25% of the Ref dataset as of release 111) than the full Ref dataset and has a more even phylogenetic distribution of sequences. We recommend this dataset to be used as the standard SILVA reference dataset for rRNA gene-based classification, phylogenetic analysis and probe design.
Living tree project
The ‘All-Species’ Living Tree Project (LTP) is a multi-partner initiative coordinated by the Journal Systematic and Applied Microbiology (Elsevier publishers) in cooperation with the ARB, LPSN and SILVA projects and promoted by the SILVA web resource. Its main objective is to provide highly curated ribosomal 16S and 23S RNA sequence datasets of all type strains representing the up-to-date described bacterial and archaeal diversity. The LTP database is kept updated according to the changes in nomenclature and new descriptions of taxa that are effectively published in the International Journal of Systematic and Evolutionary Microbiology. New type-strain sequences are carefully examined by means of their sequence quality, associated (meta) data and manual inspection of the alignment. This process results in: (i) finding the best available SSU/LSU entry that may represent a species; (ii) providing corrected organism-name information plus other LTP-specific (meta) data and (iii) propagating the alignment improvements to the SILVA seed alignment. These very small but taxonomically ‘comprehensive’ datasets are frequently used for taxonomic and classification purposes and are useful as test datasets for developers.
Further developments
Data retrieval
The semantic interpretations of ‘gene’, ‘product’ and ‘note’ feature qualifiers were modified to avoid overlapping/duplicate entries in the SILVA database due to insufficiently annotated EMBL-Bank entries.
Aligner
The SILVA database preparation pipeline now employs an updated version (1.2.10) of SINA to compute the multiple sequence alignment provided with the databases. Please refer to Pruesse et al. (10) for a detailed description of SINA. Briefly, SINA is a reference-based alignment tool, designed to maintain high alignment accuracy while allowing for volume sequence processing. For each sequence, SINA selects a set of similar sequences from the given reference alignment and constructs a directed acyclical graph (DAG) representation of the alignment of these reference sequences to be used as alignment template. The computed alignment of the query sequence and the DAG template is optimal under the constraint that no columns may be added to the alignment.
The base pair score reported by SINA is an indicator for the degree to which the expected secondary structure is met by the aligned sequence. The SILVA alignments each include a global secondary structure, defining which pairs of alignment columns are expected to bond. The SINA ‘bp score’ is calculated as the average binding score of the column pairs covered by the aligned sequence. The alignment identity (SINA version 1.2.10 and above) reports the highest fractional identity between the query sequence and any sequence in the reference alignment.
Internal reference datasets
Several reference datasets are used during database preparation. These are the alignment SEED, a vector sequence database and a collection of non-chimeric sequences. Each of these datasets was continuously improved and extended with trusted data. The vector sequence database is available for download via the website archive. The alignment SEED and the collection of non-chimeric sequences derived from the SEED, however, must remain undisclosed. While we would prefer to make this dataset available, we are not free to do so as it contains sequences obtained under the promise of confidentiality. The SEED is extended with additional sequences when a specific phylogenetic branch is found to require a more detailed alignment. The sequences leading to such findings are typically novel and entailed much effort by the respective author to ensure their validity, making it impossible for us to also ask for publication rights. However, once those sequences do become published, they will automatically appear in the next SILVA release. Obtaining the intersection between the SEED and the Ref can be done via the field ‘align_log_slv’. SINA guarantees that sequences identical to (or sub-sequences of) reference sequences retain the exact alignment of the respective reference sequence and marks the sequences accordingly. The dataset used to evaluate SINA was prepared by selecting those sequences from the Ref 108 and is available for download on the SILVA website. However, for optimal alignment and classification results we recommend to use the Ref NR dataset as reference dataset. While this dataset is about five times larger than the SEED and is not purely comprised of manually curated sequences, it has by way of its construction a relatively even sequence distribution and good phylogenetic completeness.
SILVA WEBSITE
The SILVA website comprises core database access features, several online tools and an extensive, regularly updated set of documentation pages. The documentation pages provide tutorials for all SILVA tools and functionalities, FAQs and a detailed documentation page for each released database. The website also hosts information for partner projects and collaborations such as LTP and ETWG.
Core database access features
The SILVA databases can be accessed online via the Taxonomy Browser and Search pages. The Browser implements a hierarchical view on the database contents, similar to a file browser, visualizing any of the taxonomies included with SILVA (SILVA, LTP, RDP-II, greengenes and EMBL-Bank). The Search page supports keyword matches on a variety of fields (publication details, organism name, DOI/PubMed identifiers, etc.) as well as range filters on numeric descriptors (sequence length, quality values and dates). Multiple accession numbers can be matched by pasting comma separated lists or ranges of accession numbers in the respective field. Furthermore, searches can be constrained to the Ref, Ref NR or LTP datasets and restricted to the contents of the Cart.
The Cart system connects the different tools on the SILVA website by storing a set of sequences of interest. The Cart’s contents can be modified and displayed in both the Browser and the Search. When sequences are added to the Cart, these sequences will be highlighted by the Browser and sequence counts will be shown for each displayed taxonomic group. In line with the Cart metaphor, it is also possible to have the Cart’s contents prepared for download. Export files can be generated in ARB and FASTA formats, with and without alignment and optionally compressed.
Within the Search, the Cart allows to express complex questions as a series of simple queries. For example, searching for all sequences marked as type strain by StrainInfo but not by RDP-II can be achieved by first adding all sequences that contain the strain tag ‘s[T]’ to the cart and then removing those sequences for which strain field is marked with ‘r[T]’.
Alignment, sequence-based search and classification
The Aligner page allows submitting sequence data for processing with SINA. FASTA files containing up to 1000 sequences (≤6000 nt each) can be uploaded, and the sequences will be aligned using the same reference alignments that are employed to prepare the SILVA databases. While all SINA parameters are configurable in the web form, the parameters will default to the same values that were used to prepare the most recent SILVA release.
Optionally, the aligned sequences can be passed through the search stage of SINA to find closely related sequences in the Parc, Ref or Ref NR datasets. The query sequences are compared with the sequences in the selected dataset based on the SINA alignment. The search results can then be added to the Cart, and thereby accessed from the other components of the website, for example to prepare a download or to inspect the taxonomic groups containing sequences similar to the submitted query sequences.
It is also possible to classify the submitted sequences based on the search results. For each query sequence, the classifications assigned to the matched sequences are consolidated using a lowest common ancestor approach. The resulting classification is included with the aligned sequence data.
Probe and primer evaluation
Signature sequences are essential to many methods employed in the investigation of microbial communities. Since these signatures are derived from previously characterized sequences, they can only be accurate to the degree to which diversity was covered by available data at the time of their design. As more data become available over time, signature sequences must be regularly re-evaluated and their suitability re-affirmed. In order to make this process as simple as possible, the TestProbe and TestPrime tools are now offered on the SILVA website. Users can base their calculations on the entire SSU or LSU Parc datasets, or on the Ref or Ref NR subsets. The results can be downloaded as CSV files and matched sequences can be added to the Cart for subsequent download. Both TestProbe and TestPrime are cross-linked with the oligonucleotide signature database probeBase (23).
TestProbe
The probe match and evaluation tool tests and visualizes in silico target group coverage of rRNA gene-targeting probes and single primers, optionally with ambiguity codons, against the SILVA datasets. The tool can be configured to allow up to five mismatches between the probe and target sequences. Mismatches can also be weighted. The results are shown in three tables: an overview of the number and position of mismatches, a per-taxon summary table and a third table listing individual matching sequences with sequence names, accession numbers and a graphical representation of the probe’s binding site within all matches.
TestPrime
Similar to the TestProbe tool, TestPrime allows searching for all sequences within the SILVA databases which are targeted by a pair of primers (optionally with ambiguity codons). The maximal number of allowed base mismatches can be configured, as well as a ‘zero tolerance zone’ at the 3′-end of the primers. Coverage and match, mismatch and no data information are shown in overview pie charts (Figure 1). The graphical results are complemented by two tables similar to TestProbe.
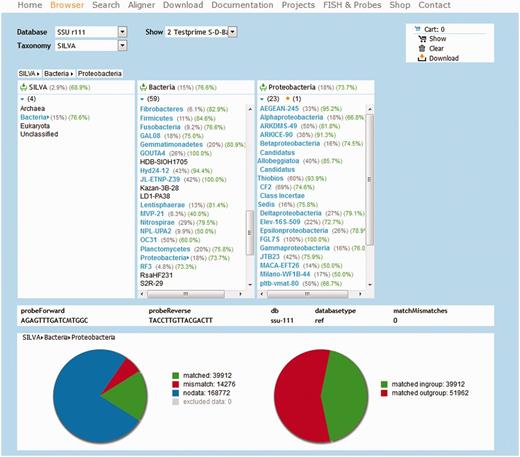
Screenshot of the Taxonomy Browser showing TestPrime results for two universal primers for evaluation.
The in silico stage of TestProbe and TestPrime is built upon the ARB ‘PT server’. First, the signature sequences are resolved into sets of ambiguity-free oligonucleotides. Each oligonucleotide is then analyzed by the PT server according to the configured stringency parameters. The results are sorted into three groups: match, mismatch and no data. The last group contains all sequences for which no clear decision could be made. Most commonly, this is the case when the sequence in question does not cover the oligonucleotide match position (short or partial sequences).
SILVA direct link API
Direct linking into the browser is supported by URLs of the form: http://www.arb-silva.de/browser/{lsu,ssu}/<INSDC accession number>.
Direct linking into the search is supported by URLs of the form: http://www.arb-silva.de/search/show/{lsu,ssu}/<search field>/<search term>[<search field>/<search term>[…]].
Up to four pairs of search fields and search terms are allowed. A description of the available search fields is documented on the website. They include ‘acc’ for INSDC accession numbers, ‘name’ for organism name and ‘pubid’ for PubMedID or DOI.
SILVA entries are already directly linked from various sources, including the EMBL-Bank, the GenBank, probeBase and StrainInfo.net.
OUTLOOK
The full impact of the ‘data deluge’ originating by the advent of NGS technologies has not yet influenced the amount of long, assembled sequence data such as full-length rRNA gene sequences. However, the growth of rRNA gene data has already rendered many comparative analysis methods impossible to be applied on comprehensive datasets.
We expect that tree reconstruction for the complete SSU Ref datasets will become infeasible in the near future. Taxonomy curation will then be based on the smaller SSU Ref NR dataset. Classifications for all other sequences in the SILVA databases will be created with a high-throughput approach using this curated Ref NR taxonomy as a reference.
Our databases are already popular in high-throughput analysis pipelines and we expect the importance of these applications to further increase in the future. We are, therefore, committed to enhancing the usability of our reference datasets for these applications. The work on our taxonomy, in particular on its eukaryotic branch, will be of direct and transparent benefit to analysis procedures relying on the SILVA databases.
FUNDING
Max Planck Society; Deutsche Forschungsgemeinschaft [GL 553/4-1]. Funding for open access charge: Deutsche Forschungsgemeinschaft.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors would like to thank Wolfgang Ludwig and Ralf Westram for expert assistance with the ARB software suite, the alignments and phylogeny. They greatly appreciate the help the SILVA users have rendered with critical evaluation and feedback on the SILVA databases and tools. They would also like to thank the RDP-II, StrainInfo and probeBase teams, as well as our taxonomy collaboration partners for their support and many fruitful discussions.
REFERENCES
Author notes
The authors wish it to be known that, in their opinion, the first three authors should be regarded as joint First Authors.

{kind=link}
Comments