
Abstract
Background:
In order to investigate the mechanisms of acquired resistance to trabectedin, trabectedin-resistant human myxoid liposarcoma (402-91/T) and ovarian carcinoma (A2780/T) cell lines were derived and characterised in vitro and in vivo.
Methods:
Resistant cell lines were obtained by repeated exposures to trabectedin. Characterisation was performed by evaluating drug sensitivity, cell cycle perturbations, DNA damage and DNA repair protein expression. In vivo experiments were performed on A2780 and A2780/T xenografts.
Results:
402-91/T and A2780/T cells were six-fold resistant to trabectedin compared with parental cells. Resistant cells were found to be hypersensitive to UV light and did not express specific proteins involved in the nucleotide excision repair (NER) pathway: XPF and ERCC1 in 402-91/T and XPG in A2780/T. NER deficiency in trabectedin-resistant cells was associated with the absence of a G2/M arrest induced by trabectedin and with enhanced sensitivity (two-fold) to platinum drugs. In A2780/T, this collateral sensitivity, confirmed in vivo, was associated with an increased formation of DNA interstrand crosslinks.
Conclusions:
Our finding that resistance to trabectedin is associated with the loss of NER function, with a consequent increased sensitivity to platinum drugs, provides the rational for sequential use of these drugs in patients who have acquired resistance to trabectedin.
Similar content being viewed by others


Main
Trabectedin is a marine-derived tetrahydroisoquinoline alkaloid with antitumour activity. Its peculiar pentacyclic structure allows the interaction (through covalent binding) with the N2-position of guanine in the minor groove of the DNA (Pommier et al, 1996; Zewail-Foote and Hurley, 2001). Once bound, trabectedin is believed to interact with DNA-binding molecules, including transcription factors and DNA repair proteins (Forni et al, 2009). This interaction affects the transcription of activated genes in a promoter- and gene-specific manner and also results in a bending of the helix towards the major groove. This distortion causes the activation of pathways involved in DNA repair. Homologous recombination (HR) is particularly important for trabectedin efficacy: indeed HR-deficient cells are approximately 100 times more sensitive to the drug. In contrast, non-homologous end joining and mismatch repair deficiency do not appear to affect the cytotoxic activity of this drug (D'Incalci and Galmarini, 2010).
A peculiar aspect of the mechanism of action of trabectedin is its pattern of activity in nucleotide excision repair (NER)-deficient cells: the drug showed decreased activity (from 2- to 10-fold) in these cells compared with NER-proficient cells. It is thought that DNA-bound trabectedin prevents the correction of DNA lesions by transcription-coupled NER (TC-NER) by creating cytotoxic ternary complexes with DNA-binding proteins of the NER system, such as XPG (Damia et al, 2001; Erba et al, 2001; Tavecchio et al, 2007). The formation of such complexes would then induce both transcription- and replication-coupled DNA double-strand breaks (DSBs) that require HR to be repaired.
Despite trabectedin having a unique and complex mechanism of action, trabectedin-treated patients frequently develop resistance to the drug. The mechanism of trabectedin resistance in vitro is not completely understood as few acquired resistant cell lines have been described. Several in vitro studies (Kanzaki et al, 2002; Shao et al, 2003) showed that prolonged exposure to trabectedin induced the downregulation of Pgp1 (multidrug resistance-associated protein) expression.
In contrast with these data, Erba et al (2000) described Pgp1 overexpression in an ovarian cancer cell line resistant to trabectedin, Igrov-1/25ET.
The study of mechanisms underlying trabectedin resistance must take into consideration that trabectedin impairs transcription regulation (Jin et al, 2000; Minuzzo et al, 2000). This ability appears to be correlated with the induction of resistance in cancer cell lines, as showed by Marchini et al (2005). They observed changes in the gene expression profile of several genes (coding for transcription factors, cytoskeleton reorganisation enzymes, signal transduction proteins and enzymes involved in cellular metabolism) between parental and trabectedin-resistant cells.
In this study, we developed a human myxoid liposarcoma and an ovarian cancer cell line (402-91/T and A2780/T, respectively) resistant to trabectedin. Based on the assumption that trabectedin efficacy is correlated to the activity of DNA repair systems and transcription regulation, we characterised the resistant cell lines with both cellular and molecular approaches. The finding that trabectedin resistance is associated with collateral sensitivity to other chemotherapeutic agents, both in vitro and in vivo systems, has potential clinical applications.
Materials and methods
Cells
In order to generate trabectedin-resistant cell lines, myxoid liposarcoma 402-91 and ovarian carcinoma A2780 cells were exposed to a stepwise increase in drug concentration using a short (1 h) exposure for 10 treatments. Cells were grown in RPMI-1640, 10% FBS and 1% L-glutamine and maintained at 37 °C in a humidified atmosphere at 5% CO2. The resistant cell lines were termed 402-91/T and A2780/T, respectively, and the resistance index was evaluated by clonogenic assay (Tavecchio et al, 2008).
Drugs
Trabectedin was kindly provided by PharmaMar, S.A. (Colmenar Viejo, Spain), stocked in DMSO at a concentration of 1 mM and stored at −20 °C. Cis-diammineplatinum (II) dichloride (cis-DDP) and carboplatin were purchased from Sigma Aldrich, St Louis, MO, USA. All drugs were diluted in RPMI-1640 medium just before use.
Proliferation assay
The evaluation of cisplatin and carboplatin cross-resistance on 402-91/T and A2780/T cell lines was assessed by WST-1 cell proliferation assay (Roche, Basel, Switzerland) (Romano et al, 2013).
Flow cytometric cell cycle analysis
Cell cycle perturbations induced by trabectedin and cis-DDP were evaluated by standard flow cytometric methods. Control and treated cells were counted by using Coulter Counter (ZM, Beckman Coulter, Brea, CA, USA) every 24 h after drug washout and fixed in 70% ethanol before DNA staining (Erba et al, 2001).
Flow cytometric γ-H2AX detection
At the end of treatment and at different times after drug washout, the cells were fixed and stained for γ-H2AX detection as previously described (Tavecchio et al, 2008).
RNA purification and semi-quantitative RT–PCR
Total RNA was purified using the RNeasy Mini Kit (Qiagen, Milan, Italy) according to the manufacturer’s instructions. Reverse transcription to cDNA was performed using a High Capacity cDNA Reverse Transcription Kit (Life Technologies, Carlsbad, CA, USA) starting from 500 ng of total RNA following the manufacturer’s instructions. Differences in ERCC1, XPF and XPG expression were determined by real-time RT-PCR (ABI-7900, Applied Biosystems, Carlsbad, CA, USA) using Sybr Green (Qiagen) and the following primers (ERCC1 FW: 5′-CCAACAGCATCATTGTGAGC-3′; ERCC1 RV: 5′-CGGGAATTACGTCGCCAAAT-3′; XPF FW: 5′-TTGTGAGGAAACTGTATCTGTGG-3′; XPF RV: 5′-AGCAAGCATGGTAGGTGTCA-3′; XPG FW: 5′-TCTGGAAGCTGCTGGAGTG-3′; XPG RV: 5′-TGCTAATATCAACAGCCAGGAT-3′). The analysis was performed using the 2−DDCT method (Livak and Schmittgen, 2001).
Western blotting analysis
Whole-protein extracts were obtained using a lysis buffer containing 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA and 1% Triton in the presence of protease inhibitors (Roche). Proteins were quantified using the Bradford assay reagent (Bio-Rad Laboratories, Melville, NY, USA), separated on a SDS–PAGE gel, transferred to a nitrocellulose membrane (Bio-Rad) and probed with the primary antibodies to XPG (Bethyl, Montgomery, TX, USA), ERCC1 (D6G6 XP, Cell Signaling Technology, Danvers, MA, USA), XPF (D3G8C, Cell Signaling Technology) and β-tubulin (H235, Santa Cruz Biotechnology, Dallas, TX, USA). Binding was detected using peroxidase-labeled secondary antibodies and visualised using a Chemiluminescence Kit (Thermo Scientific, Waltham, MA, USA).
Western blotting analysis was performed periodically (at least every 5 months) and for every new batch of cells that were defrosted.
Single-cell gel electrophoresis (comet) assay
The modified single-cell gel electrophoresis (comet) assay (Hartley et al, 2011) was used to measure DNA interstrand crosslinking (ICL) induced by platinum drug treatment in parental and resistant cells. Exponentially growing A2780 cells were treated for 1 h with different concentrations of cis-DDP and collected after 9 h. All procedures were carried out on ice. Cells were diluted to a final concentration of 2.5 × 104 cells ml−1 and irradiated with X-ray (15 Gy) in order to deliver a fixed number of random DNA strand breaks (except for the untreated unirradiated control). Cells were embedded in 1% agarose on a precoated microscope slide, and each sample was prepared in duplicate. In subdued light, cells were lysed for 1 h in lysis buffer (100 mM disodium EDTA, 2.5 M NaCl, 10 mM Tris–HCl pH 10.5 and 1% Triton X-100 added at use) and then washed four times in distilled water every 15 min. Slides were incubated in alkali buffer (50 mM NaOH, 1 mM disodium EDTA, pH12.5) for 45 min followed by electrophoresis in the same buffer for 25 min at 18 V (0.6 V cm−1), 250 mA. Finally, the slides were rinsed in neutralising buffer (0.5 M Tris–HCl, pH 7.5) then in saline and allowed to dry overnight at room temperature. Re-hydrated slides were stained with propidium iodide (2.5 μg ml−1) for 30 min, rinsed in distilled water and oven-dried. Images were visualised using a NIKON inverted microscope (Chiyoda, Tokyo, Japan) with high-pressure mercury light source, 510–560 nm excitation filter and 590 nm barrier filter at × 20 magnification. Images were captured using an on-line CCD camera and analysed using the Komet Analysis software 4.02 (Andor Technology, Belfast, UK). For each duplicate slide, 25 cells were analysed. The tail moment for each image was calculated as the product of the percentage of DNA in the comet tail and the distance between the means of the head and tail distributions (Olive et al, 1990). DNA ICL after drug treatment was expressed as the percentage decrease in tail moment compared with irradiated controls calculated by the formula:

where TMdi=tail moment of drug-treated irradiated sample; TMcu=tail moment of untreated, unirradiated control; and TMci=tail moment of untreated, irradiated control.
In vivo study
Procedures involving animals and their care were conducted in conformity with the following laws, regulations and policies governing the care and use of laboratory animals: Italian Governing Law (D.lgs 26/2014; Authorisation n.19/2008-A issued 6 March 2008 by Ministry of Health); Mario Negri Institutional Regulations and Policies providing internal authorisation for persons conducting animal experiments (Quality Management System Certificate—UNI EN ISO 9001:2008—Reg. No. 6121); the NIH Guide for the Care and Use of Laboratory Animals (2011 edition); EU directives and guidelines (EEC Council Directive 2010/63/UE); and in line with Guidelines for the welfare and use of animals in cancer research (Workman et al, 2010).
Animal experiments has been reviewed and approved by the IRFMN Animal Care and Use Committee (IACUC) that includes members ‘ad hoc’ for ethical issues. Animals were housed in the Institute’s Animal Care Facilities, which meet international standards; they were regularly checked by a certified veterinarian who is responsible for health monitoring, animal welfare supervision, experimental protocols and procedure revision.
For in vivo studies, 4–6-week-old female athymic nude mice (Harlan Laboratories, Milan, Italy) maintained under specific pathogen-free conditions were used. Human ovarian A2780 cancer cells (10 × 106 cell per mouse) were implanted s.c. into the flank of recipient mice. When tumour was palpable (mean tumour weight value for the groups was about 100 mg), animals were divided randomly into treatment groups consisting at least of eight mice each. Tumour growth was measured three times a week using a caliper, and the tumour weights were calculated by the formula: length × (width)2/2. Antitumour activity was expressed as T/C%, where T and C were the mean tumour weights of treated and control groups, respectively. Treatment was considered active when T/C<42%.
Trabectedin, cisplatin and carboplatin were administered intravenously once a week for three consecutive weeks (q7dx3) at the doses of 0.15, 5 and 50 mg kg−1, respectively. We used these doses and this schedule as they are the well-tolerated commonly used doses in mice and they are very close to the MTD (maximum tolerated dose) (Romano et al, 2013; Ricci et al, 2014).
Results
Trabectedin-resistant cell line derivation and characterisation
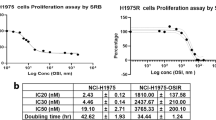
In order to investigate the mechanism of trabectedin resistance, we derived two trabectedin-resistant cell lines: 402-91/T and A2780/T. These sublines were generated from a myxoid liposarcoma and an ovarian cancer cell line, respectively, that were exposed to stepwise increasing concentrations of trabectedin for 10 1-h treatments. The sensitivity to trabectedin of the parental and resistant cell lines was tested by clonogenic assay as shown in Figure 1. We first considered the parental cell lines and observed that 402-91 cells are more sensitive to trabectedin than A2780 cells, with IC50 values 1.5 and 8.2 nM, respectively. Second, we compared the IC50 values between parental and resistant cells and we revealed that 402-91/T cells (Figure 1A) and A2780/T cells (Figure 1B) were six-fold less sensitive to trabectedin than the respective parental cell lines (1.5 vs 8.3 nM and 8.2 vs 48 nM, respectively; P⩽0.0001, Student’s t-test). For both cell lines, the resistance was irreversible over 1 year of continuous culture in drug-free medium, and no changes in morphology and growth features, compared with the parental cell lines, were observed.

Sensitivity to trabectedin of the parental and resistant cell line tested by clonogenic assay. (A) 402-91 (•) vs 402-91/T (Δ); (B) A2780 (•) vs A2780/T (Δ). Each point is the mean of five replicates of three independent experiments; bars represent s.d.
Sensitivity to UV and involvement of NER
As demonstrated in previous studies (Takebayashi et al, 2001; Uboldi et al, 2012), trabectedin resistance is frequently associated with defects in the NER pathway and with increased sensitivity to UV light. We analysed the effects induced by UV light exposure in the resistant and parental cell lines. Figures 2A and B showed that both the 402-91/T and A2780/T cells were more sensitive (four- and six-fold, respectively) to UV light than the respective parental cells.

Effect of UV rays on the clonogenicity of (A) 402-91 (•) and 402-91/T (Δ) cell lines; (B) A2780 (•) and A2780/T (Δ) cell lines. Each point is the mean of five replicates of three independent experiments; bars represent s.d. Western blotting analysis of ERCC1, XPF and XPG protein expression in (C) 402-91 and 402-91/T; (D) A2780 and A2780/T.
The high sensitivity of the resistant cell lines to UV light prompted us to investigate the expression of key proteins of the NER pathway, including XPG, XPF and ERCC1 (Figures 2C and D). In the A2780/T cell line, western blotting analysis revealed no detectable XPG expression, while RT–PCR analysis showed a slight downregulation of mRNA expression (fold change 0.43) when compared with parental cells. In contrast, 402-91/T did not express XPF and ERCC1, but no differences were observed in mRNA level for both genes (fold change 0.70 and 0.84, respectively).
Cell cycle effects and platinum drug sensitivity
In order to better characterise the resistant cell lines, we analysed the cell cycle perturbations induced by trabectedin using standard flow cytometric methods. DNA cell cycle analysis was performed on controls and cells treated for 1 h with different concentrations of trabectedin and collected 24, 48 or 72 h after drug washout. As shown in Supplementary Figures S1A and C, trabectedin caused accumulation of cells in G2/M phase in 402-91 at 2.5 nM and in A2780 at 5 nM, already detectable at 24 h. The analysis performed at 48 and 72 h demonstrated that the cells overcame the block and progressed through the cell cycle. In contrast, the cells treated with a higher drug concentration remained blocked in the G2/M phase of the cell cycle up to 72 h. In resistant cell lines, trabectedin did not induce the G2/M block, even at higher drug concentrations (Supplementary Figures S1B and D). These data were in agreement with previous studies showing that NER status affected trabectedin efficacy: NER-proficient cells were more sensitive to the drug than NER-deficient ones (Damia et al, 2001) and displayed different perturbations of the cell cycle consisting of a greater accumulation of cells in G2/M phases after trabectedin treatment (Tavecchio et al, 2007).
Previous studies have demonstrated that cell lines with NER deficiencies were more sensitive to cisplatin treatment (Stevens et al, 2008; Wood, 2010). As shown in Figure 3, we evaluated the sensitivity of the trabectedin-resistant and parental cell lines to platinum drugs. We treated the cells with cisplatin (1-h treatment) or carboplatin (24-h treatment) and observed, for both cancer cell lines, that trabectedin-resistant sublines were two-fold more sensitive to platinum drugs than the parental lines.

Collateral sensitivity to cisplatin or carboplatin assessed by WST-1 cell proliferation assay: (A and C) 402-91 (•) vs 402-91/T (Δ); (B and D) A2780 (•) vs A2780/T. Each point is the mean of five replicates of three independent experiments; bars represent s.d.
γ-H2AX DNA damage response
The formation of phophorylated histone H2AX (γ-H2AX) is considered a sensitive marker for the detection of DSBs (Dickey et al, 2009). As shown in previous studies (Soares et al, 2005; Guirouilh-Barbat et al, 2008; Tavecchio et al, 2008) trabectedin induces replication and transcription-coupled DSBs whose formation can be influenced by NER activity. H2AX phophorylation can also act as a sensitive and general marker of DNA damage induced by cisplatin in several cell models (Clingen et al, 2008; Spanswick et al, 2012). To measure possible differences in γ-H2AX formation between the A2780 and A2780/T cell lines, we performed flow cytometric γ-H2AX analysis after trabectedin or cisplatin treatment. As shown in Figure 4B, treatment with equitoxic doses of trabectedin (20 nM for A2780 and 80 nM for A2780/T) generated a higher percentage of γ-H2AX-positive cells in the A2780 cell line. The induction of this DNA damage marker was time dependent in both cell lines but the distribution in the cell cycle phases of γ-H2AX-positive cells was different (Figure 4A). Starting from 8 h after drug washout, A2780-damaged cells were principally in early S phase while in A2780/T positive cells were detected in all the cell cycle phases with an accumulation in late S–G2/M phase. Figure 4D shows that induction of H2AX phophorylation after treatment with equitoxic doses of cis-DDP (30 μ M in A2780 and 15 μ M in A2780/T) was time dependent and comparable for both cell lines. The analysis of the cell cycle phase distribution of cis-DDP-treated cells (Figure 4C) revealed that 24 h after drug washout the γ-H2AX-positive cells were principally accumulated in S–G2/M phases both in A2780 and A2780/T cell lines.

Analysis of γ-H2AX levels in A2780 and A2780/T cell lines after 1 h (A and B) trabectedin and (C and D) cisplatin treatment at different time points: 1hT (treatment), 2, 4, 8, and 24hR (drug washout). Each point is the mean of three independent experiments; bars represent s.d.
DNA interstrand crosslinking
Spanswick et al (2012) demonstrated that A2780 cells were not able to ‘unhook’ cisplatin-induced DNA ICLs from DNA over a 48-h period. For this reason, we performed a modified comet assay to compare the levels of ICL formation after cisplatin treatment in the parental and trabectedin-resistant cell lines. Cells were treated for 1 h followed by 9-h incubation in drug-free medium to allow the peak of ICL formation, as previously described (Spanswick et al, 2012). As shown in Figure 5, treatment with equimolar doses of drug produced a higher level of ICL formation in trabectedin-resistant cells than in the parental ones.

DNA ICL formation in A2780 (•) and A2780/T (Δ) cells following treatment with cisplatin. Samples were taken at 9 h after drug washout and ICLs were measured using the modified comet assay. Data are the mean±s.d. from at least three independent experiments. *P⩽0.5; **P⩽0.01, Student’s t-test.
In vivo collateral sensitivity to platinum drugs
We performed in vivo experiments only for the ovarian cancer cell lines as the s.c. implantation of 402-91 cells did not generate an appreciable tumour mass. The collateral sensitivity to platinum drugs observed in vitro was confirmed in vivo (Figure 6). In spite of the fact that in vivo A2780 xenografts were unexpectedly found to be only marginally sensitive to trabectedin – possibly owing to pharmacokinetic reasons – A2780/T xenografts displayed a marked increase in sensitivity to cisplatin (T/C 23%) and carboplatin treatment (T/C 40%) compared with those obtained from parental xenografts (T/C: 61% and 60%, respectively).

In vivo antitumour activity of trabectedin, cisplatin and carboplatin administered intravenously q7dx3 at the doses of 0.15, 5 and 50 mg kg−1, respectively, in (A) A2780 and (B) A2780/T. *P⩽0.5, **P⩽0.01; ***P⩽0.001; ****P⩽0.0001, ANOVA test.
Discussion
In this paper, we report the development and characterisation of two trabectedin-resistant cell lines, myxoid liposarcoma 402-91/T and ovarian A2780/T cells that were six-fold less sensitive to the drug. Only a few cell lines have been described that show specific acquired resistance to trabectedin (Erba et al, 2001; Kanzaki et al, 2002; Manara et al, 2005; Marchini et al, 2005; Duan et al, 2009; Uboldi et al, 2012).
402-91/T and A2780/T cells were hypersensitive to UV radiation, and we demonstrated that both cell lines had defects in the NER pathway because they lack the expression of key proteins of this repair mechanism: XPF and ERCC1 in 402-91/T and XPG in A2780/T. These data are in keeping with previous studies conducted on cell lines with defined defects of DNA repair mechanisms and which suggested that trabectedin efficacy can be affected by NER and HR activity (Damia et al, 2001; Erba et al, 2001). Defects in HR were associated with high sensitivity to the drug while cells that are defective in TC-NER are partially resistant to trabectedin and hypersensitive to UV light (Damia et al, 2001; Takebayashi et al, 2001). The role of XPG status in trabectedin efficacy is important as this protein is part of the ternary complex (trabectedin–DNA–XPG) that stalls the DNA replication fork during S-phase resulting in DNA breaks that need to be repaired by HR (Herrero et al, 2006). It is known that XPF and ERCC1 take part in a subsequent step in removing trabectedin from the DNA (de Laat et al, 1999) and the fact that 402-91/T cells were resistant to trabectedin confirms the importance of the NER pathway in the modulation of trabectedin efficacy. Even if the relationship between transcript and protein expression is unknown for NER molecules (Stevens et al, 2008), our RT–PCR data revealed no downregulation in mRNA expression of XPF, ERCC1 and XPG, suggesting that the lack of protein expression might be due to modification at the posttranscriptional level.
Differently from the mode of action of other alkylating agents, trabectedin causes strong perturbations of the cell cycle causing a delay of cells progressing from G1 to G2 and ultimately resulting in a G2/M block in cells with functional NER (Erba et al, 2001; Minuzzo et al, 2005; Tavecchio et al, 2007). This paper supports this observation showing that trabectedin-resistant cells, which are defective for NER pathway, did not undergo the G2/M block observed in parental cells (NER proficient).
As expected, cells deficient in NER were hypersensitive to UV light and particularly sensitive to platinum drugs. In the present paper, we show for the first time that trabectedin-resistant cells exposed to platinum drugs are more sensitive to these drugs because of an increased formation of DNA ICLs that represent the critical toxic lesion of the drugs. This finding was convincingly reproduced in vivo and thus could be potentially clinically relevant. It is in fact plausible that ovarian cancer patients receiving trabectedin may become more sensitive to subsequent platinum-based therapies. This hypothesis is supported by the retrospective analysis of the survival of patients who had participated in the OVA301 trial that was designed to compare the effectiveness of the combination of trabectedin and pegylated liposomal doxorubicin (PLD) with PLD alone (Monk et al, 2015). It was found that patients who received platinum-based therapy at relapse following trabectedin survived much longer than those who received the same therapy after PLD alone (27.7 vs 18.7 months) (Poveda et al, 2014). Although this retrospective analysis requires prospective validation, the data shown in the present study provide a clear molecular rationale for the efficacy of the sequential treatment with trabectedin and platinum drugs.
Change history
22 December 2015
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Clingen PH, Wu JY, Miller J, Mistry N, Chin F, Wynne P, Prise KM, Hartley JA (2008) Histone H2AX phosphorylation as a molecular pharmacological marker for DNA interstrand crosslink cancer chemotherapy. Biochem Pharmacol 76: 19–27.
D'Incalci M, Galmarini CM (2010) A review of trabectedin (ET-743): a unique mechanism of action. Mol Cancer Ther 9: 2157–2163.
Damia G, Silvestri S, Carrassa L, Filiberti L, Faircloth GT, Liberi G, Foiani M, D'Incalci M (2001) Unique pattern of ET-743 activity in different cellular systems with defined deficiencies in DNA-repair pathways. Int J Cancer 92: 583–588.
de Laat WL, Jaspers NG, Hoeijmakers JH (1999) Molecular mechanism of nucleotide excision repair. Genes Dev 13: 768–785.
Dickey JS, Redon CE, Nakamura AJ, Baird BJ, Sedelnikova OA, Bonner WM (2009) H2AX: functional roles and potential applications. Chromosoma 118: 683–692.
Duan Z, Choy E, Harmon D, Yang C, Ryu K, Schwab J, Mankin H, Hornicek FJ (2009) ZNF93 increases resistance to ET-743 (Trabectedin; Yondelis) and PM00104 (Zalypsis) in human cancer cell lines. PLoS One 4: e6967.
Erba E, Bergamaschi D, Bassano L, Damia G, Ronzoni S, Faircloth GT, D'Incalci M (2001) Ecteinascidin-743 (ET-743), a natural marine compound, with a unique mechanism of action. Eur J Cancer 37: 97–105.
Erba E, Bergamaschi D, Bassano L, Ronzoni S, Di Liberti G, Muradore I, Vignati S, Faircloth G, Jimeno J, D'Incalci M (2000) Isolation and characterization of an IGROV-1 human ovarian cancer cell line made resistant to Ecteinascidin-743 (ET-743). Br J Cancer 82: 1732–1739.
Forni C, Minuzzo M, Virdis E, Tamborini E, Simone M, Tavecchio M, Erba E, Grosso F, Gronchi A, Aman P, Casali P, D'Incalci M, Pilotti S, Mantovani R (2009) Trabectedin (ET-743) promotes differentiation in myxoid liposarcoma tumors. Mol Cancer Ther 8: 449–457.
Guirouilh-Barbat J, Redon C, Pommier Y (2008) Transcription-coupled DNA double-strand breaks are mediated via the nucleotide excision repair and the Mre11-Rad50-Nbs1 complex. Mol Biol Cell 19: 3969–3981.
Hartley JM, Spanswick VJ, Hartley JA (2011) Measurement of DNA damage in individual cells using the single cell gel electrophoresis (Comet) assay. Methods Mol Biol 731: 309–320.
Herrero AB, Martin-Castellanos C, Marco E, Gago F, Moreno S (2006) Cross-talk between nucleotide excision and homologous recombination DNA repair pathways in the mechanism of action of antitumor trabectedin. Cancer Res 66: 8155–8162.
Jin S, Gorfajn B, Faircloth G, Scotto KW (2000) Ecteinascidin 743, a transcription-targeted chemotherapeutic that inhibits MDR1 activation. Proc Natl Acad Sci USA 97: 6775–6779.
Kanzaki A, Takebayashi Y, Ren XQ, Miyashita H, Mori S, Akiyama S, Pommier Y (2002) Overcoming multidrug drug resistance in P-glycoprotein/MDR1-overexpressing cell lines by ecteinascidin 743. Mol Cancer Ther 1: 1327–1334.
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402–408.
Manara MC, Perdichizzi S, Serra M, Pierini R, Benini S, Hattinger CM, Astolfi A, Bagnati R, D'Incalci M, Picci P, Scotlandi K (2005) The molecular mechanisms responsible for resistance to ET-743 (Trabectidin; Yondelis) in the Ewing's sarcoma cell line, TC-71. Int J Oncol 27: 1605–1616.
Marchini S, Marrazzo E, Bonomi R, Chiorino G, Zaffaroni M, Weissbach L, Hornicek FJ, Broggini M, Faircloth GT, D'Incalci M (2005) Molecular characterisation of two human cancer cell lines selected in vitro for their chemotherapeutic drug resistance to ET-743. Eur J Cancer 41: 323–333.
Minuzzo M, Ceribelli M, Pitarque-Marti M, Borrelli S, Erba E, DiSilvio A, D'Incalci M, Mantovani R (2005) Selective effects of the anticancer drug Yondelis (ET-743) on cell-cycle promoters. Mol Pharmacol 68: 1496–1503.
Minuzzo M, Marchini S, Broggini M, Faircloth G, D'Incalci M, Mantovani R (2000) Interference of transcriptional activation by the antineoplastic drug ecteinascidin-743. Proc Natl Acad Sci USA 97: 6780–6784.
Monk BJ, Ghatage P, Parekh T, Henitz E, Knoblauch R, Matos-Pita AS, Nieto A, Park YC, Cheng PS, Li W, Favis R, Ricci D, Poveda A (2015) Effect of BRCA1 and XPG mutations on treatment response to trabectedin and pegylated liposomal doxorubicin in patients with advanced ovarian cancer: exploratory analysis of the phase 3 OVA-301 study. Ann Oncol 26: 914–920.
Olive PL, Banath JP, Durand RE (1990) Heterogeneity in radiation-induced DNA damage and repair in tumor and normal cells measured using the "comet" assay. Radiat Res 122: 86–94.
Pommier Y, Kohlhagen G, Bailly C, Waring M, Mazumder A, Kohn KW (1996) DNA sequence- and structure-selective alkylation of guanine N2 in the DNA minor groove by ecteinascidin 743, a potent antitumor compound from the Caribbean tunicate Ecteinascidia turbinata. Biochemistry 35: 13303–13309.
Poveda A, Ray-Coquard I, Romero I, Lopez-Guerrero JA, Colombo N (2014) Emerging treatment strategies in recurrent platinum-sensitive ovarian cancer: focus on trabectedin. Cancer Treat Rev 40: 366–375.
Ricci F, Bizzaro F, Cesca M, Guffanti F, Ganzinelli M, Decio A, Ghilardi C, Perego P, Fruscio R, Buda A, Milani R, Ostano P, Chiorino G, Bani MR, Damia G, Giavazzi R (2014) Patient-derived ovarian tumor xenografts recapitulate human clinicopathology and genetic alterations. Cancer Res 74: 6980–6990.
Romano M, Frapolli R, Zangarini M, Bello E, Porcu L, Galmarini CM, Garcia-Fernandez LF, Cuevas C, Allavena P, Erba E, D'Incalci M (2013) Comparison of in vitro and in vivo biological effects of trabectedin, lurbinectedin (PM01183) and Zalypsis(R) (PM00104). Int J Cancer 133: 2024–2033.
Shao L, Kasanov J, Hornicek FJ, Morii T, Fondren G, Weissbach L (2003) Ecteinascidin-743 drug resistance in sarcoma cells: transcriptional and cellular alterations. Biochem Pharmacol 66: 2381–2395.
Soares DG, Poletto NP, Bonatto D, Salvador M, Schwartsmann G, Henriques JA (2005) Low cytotoxicity of ecteinascidin 743 in yeast lacking the major endonucleolytic enzymes of base and nucleotide excision repair pathways. Biochem Pharmacol 70: 59–69.
Spanswick VJ, Lowe HL, Newton C, Bingham JP, Bagnobianchi A, Kiakos K, Craddock C, Ledermann JA, Hochhauser D, Hartley JA (2012) Evidence for different mechanisms of 'unhooking' for melphalan and cisplatin-induced DNA interstrand cross-links in vitro and in clinical acquired resistant tumour samples. BMC Cancer 12: 436.
Stevens EV, Nishizuka S, Antony S, Reimers M, Varma S, Young L, Munson PJ, Weinstein JN, Kohn EC, Pommier Y (2008) Predicting cisplatin and trabectedin drug sensitivity in ovarian and colon cancers. Mol Cancer Ther 7: 10–18.
Takebayashi Y, Pourquier P, Zimonjic DB, Nakayama K, Emmert S, Ueda T, Urasaki Y, Kanzaki A, Akiyama SI, Popescu N, Kraemer KH, Pommier Y (2001) Antiproliferative activity of ecteinascidin 743 is dependent upon transcription-coupled nucleotide-excision repair. Nat Med 7: 961–966.
Tavecchio M, Natoli C, Ubezio P, Erba E, D'Incalci M (2007) Dynamics of cell cycle phase perturbations by trabectedin (ET-743) in nucleotide excision repair (NER)-deficient and NER-proficient cells, unravelled by a novel mathematical simulation approach. Cell Prolif 40: 885–904.
Tavecchio M, Simone M, Erba E, Chiolo I, Liberi G, Foiani M, D'Incalci M, Damia G (2008) Role of homologous recombination in trabectedin-induced DNA damage. Eur J Cancer 44: 609–618.
Uboldi S, Bernasconi S, Romano M, Marchini S, Fuso Nerini I, Damia G, Ganzinelli M, Marangon E, Sala F, Clivio L, Chiorino G, Di Giandomenico S, Rocchi M, Capozzi O, Margison GP, Watson AJ, Caccuri AM, Pastore A, Fossati A, Mantovani R, Grosso F, Tercero JC, Erba E, D'Incalci M (2012) Characterization of a new trabectedin-resistant myxoid liposarcoma cell line that shows collateral sensitivity to methylating agents. Int J Cancer 131: 59–69.
Wood RD (2010) Mammalian nucleotide excision repair proteins and interstrand crosslink repair. Environ Mol Mutagen 51: 520–526.
Workman P, Aboagye EO, Balkwill F, Balmain A, Bruder G, Chaplin DJ, Double JA, Everitt J, Farningham DA, Glennie MJ, Kelland LR, Robinson V, Stratford IJ, Tozer GM, Watson S, Wedge SR, Eccles SA (2010) Guidelines for the welfare and use of animals in cancer research. Br J Cancer 102: 1555–1577.
Zewail-Foote M, Hurley LH (2001) Differential rates of reversibility of ecteinascidin 743-DNA covalent adducts from different sequences lead to migration to favored bonding sites. J Am Chem Soc 123: 6485–6495.
Acknowledgements
This work was supported by AIRC 6595 to MD’I. JAH acknowledges programme grant support from CRUK (C2259/A16569). SU has received a fellowship from FIRC 13743. We thank Helen Valentine for her help with the comet assay.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Colmegna, B., Uboldi, S., Frapolli, R. et al. Increased sensitivity to platinum drugs of cancer cells with acquired resistance to trabectedin. Br J Cancer 113, 1687–1693 (2015). https://doi.org/10.1038/bjc.2015.407
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2015.407
Keywords
This article is cited by
-
INOVATYON/ ENGOT-ov5 study: Randomized phase III international study comparing trabectedin/pegylated liposomal doxorubicin (PLD) followed by platinum at progression vs carboplatin/PLD in patients with recurrent ovarian cancer progressing within 6-12 months after last platinum line
British Journal of Cancer (2023)
-
ERCC1 rs11615 polymorphism and chemosensitivity to platinum drugs in patients with ovarian cancer: a systematic review and meta-analysis
Journal of Ovarian Research (2021)
-
The diagnostic value of DNA repair gene in breast cancer metastasis
Scientific Reports (2020)
-
Functional and transcriptomic characterization of carboplatin-resistant A2780 ovarian cancer cell line
Biological Research (2019)
-
Establishment and characterisation of a new patient-derived model of myxoid liposarcoma with acquired resistance to trabectedin
British Journal of Cancer (2019)
