
Abstract
Gestational trophoblastic disease (GTD) is a group of conditions that originate from the abnormal hyperproliferation of trophoblastic cells, which derive from the trophectoderm, the outer layer of the blastocyst that would normally develop into the placenta during pregnancy. GTDs encompass hydatidiform mole (HM) (complete and partial), invasive mole, gestational choriocarcinoma, placental-site trophoblastic tumor, and epithelioid trophoblastic tumor. Of these, the most common is HM, and it is the only one that has been reported to recur in the same patients from independent pregnancies, which indicates the patients’ genetic predisposition. In addition, HM is the only GTD that segregates in families according to Mendel’s laws of heredity, which made it possible to use rare familial cases of recurrent HMs (RHMs) to identify two maternal-effect genes, NLRP7 and KHDC3L, responsible for this condition. Here, we recapitulate current knowledge about RHMs and conclude with the role and benefits of testing patients for mutations in the known genes.
Similar content being viewed by others

Introduction
Hydatidiform mole (HM) is an aberrant human pregnancy with abnormal embryonic development. It occurs once in every 600 pregnancies in Western countries [1] but at higher rates in the Middle East, Latin America, Africa, and the Far East [2–4]. Sporadic moles have a multifactorial etiology involving various combinations of several environmental and genetic factors. Among women with one HM, 10 % to 20 % have other forms of reproductive loss, mainly as spontaneous abortions [5–8]. Because the frequency of two reproductive losses (one HM and one spontaneous abortion) in these patients (10–20 %) is 2–4 times higher than the frequency of two spontaneous abortions in the general population (2–5 %) [9–11], it is believed that some of these patients have genetic predisposition to recurrent reproductive loss.
Recurrent hydatidiform moles are defined by the occurrence of at least two molar pregnancies in the same patient. The earliest report of RHMs available to us through PubMed search is by Mack and Catherwood in 1930 [12]. In this paper, the authors describe one case of 10 RHMs, review the literature for cases of RHMs, and cite a report in 1912 by Essen-Moeller of a patient with 18 RHMs. The frequency of RHMs varies among populations and countries. In Western countries, studies by several groups from various countries have shown that 1 % to 2 % of patients with a prior mole have a second one [5, 13, 14]. However, higher frequencies of RHMs are reported from the Middle and Far East; in these regions, the frequency of RHMs ranges from 2.5 % up to 9.4 % [6, 15–18]. In rare cases, RHMs have been seen in related women from the same family, and these cases are termed familial cases of RHMs. Such cases are considered very rare, and their frequency is not known.
At the clinical level, patients with RHMs do not have any particular feature that distinguishes them from those with nonrecurrent sporadic moles [19], which highlights the importance of DNA testing to determine patients who are at risk for mole recurrence. Also, current data indicate that determining the parental contribution to the molar tissues can help detect patients at higher risk for mole recurrence. In this review, we summarize known data about RHMs and highlight the benefits of DNA testing.
NLRP7
NLRP7, a nucleotide oligomerization domain (NOD)-like receptor, pyrin containing 7, maps to 19q13.4 and is the first identified causative gene for RHMs [20]. Studies from various groups and populations concur that NLRP7 is a major gene for this condition and is mutated in 48–80 % of patients with at least two HMs, depending on patients’ ascertainment criteria and populations [21–24]. To date, 47 mutations in NLRP7 have been reported in patients with two defective alleles (Fig. 1a) ([25] and http://fmf.igh.cnrs.fr/ISSAID/infevers/). These mutations include stop codons, small deletions or insertions (less than 20-bp), splice mutations, large deletions or insertions, and complex rearrangements. In addition to these mutations, two protein-truncating mutations, a stop codon, L823X [21], and a deletion of 60-kb extending from intron 8 of NLRP7 to intron 11 of NLRP2 [26] and approximately 17 missenses have also been seen as single heterozygous mutations or variants in patients with recurrent and sporadic moles (Fig. 1a) [26–31]. However, the pathological significance of these single mutations or variants is still the subject of debate, and more data are needed to reach a conclusion on their potential involvement in the causation or genetic susceptibility for moles. NLRP7 transcripts have been identified in several human tissues, including endometrium, placenta, hematopoietic cells, all oocytes stages, and preimplantation embryos. NLRP7 transcripts decrease after fertilization and during preimplantation development to reach their lowest level at day 3 of embryonic development, which corresponds to the blastocyst stage, and then increase sharply from day 3 to day 5, which coincides with the transcriptional activation of the embryonic genome.

Schematic representations of NLRP7 and KHDC3L protein structures with identified mutations and non-synonymous variants in patients with hydatidiform moles and reproductive loss. (a) NLRP7 protein structure with its domains. PYD = pyrin domain; NACHT == domain present in NAIP, CIITA, HET-E, and TP1 family proteins; ATP = 5′-triphosphate binding motif; LRR = leucine-rich repeats. The ATP binding domain is a small motif of 8 amino acids and starts at position 178. (b) KHDC3L protein structure with identified mutations and non-synonymous variants. KH stands for K homology domain. Mutation nomenclature is according to the Human Genome Variation Society guidelines (http://www.hgvs.org/mutnomen/recs.html). Mutations found in patients with two defective alleles are in red. Non-synonymous variants (NSVs) found only in patients in heterozygous state and not in controls are in blue. NSVs found in patients and in subjects from the general population are in black. Mutations found in patients who had at least one live birth are underlined
Functional Roles of NLRP7
NLRP7 codes for 1037 amino acid protein (including all coding exons of all splice isoforms) and has three main domains: pyrin, NACHT (i.e., found in the NAIP, CIITA, HET-E, and TP1 family proteins) and 10 leucine-rich repeats (LRR). NLRP7 is a member of the NLR family of proteins with a role in inflammation and apoptosis. Below, we outline known roles of NLRP7 in various cellular models and discuss their potential involvement in the pathophysiology of recurrent moles.
Overexpressed NLRP7 Downregulates Intracellular IL-1β
Emerging data from three different groups about the role of NLRP7 indicate that its overexpression in transient transfections downregulates the production of IL-1β, an important mediator of the inflammatory response. The first study by Kinoshita et al. demonstrated that overexpressed NLRP7 interacts with overexpressed pro-IL-1β and pro-caspase-1 and downregulates caspase-1-dependent IL-1β secretion in HEK293 cells by inhibiting pro-IL-1β processing [32]. Another study by Messaed et al. confirmed the inhibitory effect of overexpressed NLRP7 on IL-1β, but showed that NLRP7 acts primarily on pro-IL-1β and inhibits its intracellular synthesis [33•]. In addition, this study showed that NLRP7 inhibitory function is mediated concomitantly by its three domains, and mostly by the LRR. Although the precise mechanism by which NLRP7 downregulates intracellular IL-1β (pro- or mature) is not fully understood, NLRP7 has been shown to interact physically with IL-1β, caspase-1, and ASC, with the latter mediated by the pyrin domain [32, 34].
Physiological Level of NLRP7 Inhibits IL-1β Secretion in Monocytic Cells
Using an ex vivo cellular model, Messaed et al. also looked at the consequences of NLRP7 mutations on IL-1β secretion by peripheral blood mononuclear cells (PBMCs) from patients with NLRP7 mutations [33•]. They showed that patient cells secrete lower levels of IL-1β than control cells despite the fact that these same cells have normal or slightly higher amounts of intracellular pro-IL-1β synthesis, indicating NLRP7’s role in IL-1β secretion into the extracellular milieu. These findings are in line with those obtained by Kinoshita et al. in stable transfections of THP-1 cells (of human monocytic origin), where expressing an N-terminal 35-kDa NLPR7 fragment, which mimics some protein-truncating mutations observed in patients with RHMs, reduced IL-1β secretion. This finding was also confirmed in a third cellular model described by Khare et al., who demonstrated that NLRP7 knockdown using small interfering RNA in macrophages significantly impairs IL-1β release upon stimulation with microbial acylated lipopeptides [34]. Within monocytic cells, NLRP7 co-localizes with the Golgi and microtubule-organizing center and associates with microtubules. This suggests that NLRP7 mutations may decrease cytokine secretion by affecting the structure of cytoskeletal microtubules, either directly or indirectly, and impairing the trafficking of IL-1β-containing vesicles [33•]. This suggestion is further supported by the fact that treating hematopoietic cells with nocodazole, a microtubule depolymerizing agent, fragmented NLRP7’s signal [33•].
An Interesting Emerging Role for NLRP7 in Trophoblast Differentiation
Another novel and interesting role for NLRP7 was recently demonstrated by Mahadevan et al. In this study, the authors showed that NLRP7 knockdown in human embryonic stem cells led to an earlier expression of two trophoblast differentiation markers, GCM1 and INSL4, suggesting that NLRP7 loss of function accelerates trophoblast differentiation [35•]. Another interesting finding in this study was that NLRP7 knockdown increased the level of human chorionic gonadotropin (hCG), known to be very high in patients with molar pregnancies. This new role of NLRP7 is very important in view of the fact that hydatidiform mole is characterized by hyperproliferation of the trophoblast and production of high levels of hCG.
Possible Roles of NLRP7 in the Pathology of Moles
The known functions of NLRP7 in inflammatory signalling of hematopoietic cells raise questions as to whether NLRP7’s role in IL-1β production may be the cause of the early embryonic development arrest observed in molar pregnancies. Available data do indicate some connection between IL-1β, ovulation, and oocyte maturation. For instance, in several mammalian species, intra-follicular injection of IL-1β increases the rate of ovulation, but decreases the quality of the oocytes and, consequently, the rate of normal embryonic development [36, 37]. However, this role for IL-1β in oocytes is in contradiction with data on cells from patients with NLRP7 mutations, which secrete lower amounts of IL-1β. In addition, mice lacking IL-1β [38] or Type 1 IL-1 receptor (Il1r1) [39] are fertile, indicating that the lack of IL-1β signalling does not significantly affect fertility and embryo viability in mice. In addition to its role in IL-1β secretion, NLRP7 has been shown to promote cellular proliferation and invasion in testicular and endometrial cancer, respectively.
In conclusion, we believe that, individually, none of the above-described roles of NLRP7 may explain the pathology of moles or recapitulate all of their features, as it is impossible to model a pregnancy in any cellular assay. Perhaps a combination of the above-described functions, with some acting in the oocytes and affecting the differentiation and proliferation of embryonic and trophoblastic tissues, and others acting in hematopoietic inflammatory cells present in the endometrium and downregulating the maternal immune response, together contribute to the three fundamental aspects of moles: retained human pregnancies with no embryo and excessive trophoblastic proliferation. We believe that the role of NLRP7 in downregulating maternal inflammation (intra- or extracellular) and the inability of patients to spontaneously eliminate these unviable pregnancies is a fundamental aspect of this disease that distinguishes it from all other forms of early foetal loss. Indeed, it is the retention of these early arrested pregnancies that has homogenized and distinguished this category of foetal loss from all other forms of early spontaneous abortions and has consequently facilitated the identification of two of its causative genes.
KHDC3L
KHDC3L (KH domain containing 3-like), which was identified in 2011, is a second recessive gene responsible for RHMs [40•]. KHDC3L maps to chromosome 6, and available data indicate that this gene is a minor gene for RHMs, accounting for 10–14 % of patients who do not have mutations in NLRP7. To date, four mutations in KHDC3L have been reported in patients with two defective alleles (Fig. 1b) [40•, 41]. KHDC3L transcripts have been identified in several human tissues, including all oocytes stages, preimplantation embryos, and hematopoietic cells. KHDC3L codes for a small protein of 217 amino acids belonging to the KHDC1 (KH homology domain containing 1) protein family, members of which contain an atypical KH domain that does not bind RNA as opposed to proteins with canonical KH domain. In humans, this family includes KHDC3L, KHDC1, DPPA5 (developmental pluripotency associated 5), and OOEP (oocyte-expressed protein) [42]. Expression of KHDC3L is highest in oocytes at the germinal vesicle stage and then decreases during preimplantation development and becomes undetectable at the blastocyst stage [40•], similar to the expression prolife of NLRP7 [43]. In addition, KHDC3L co-localizes with NLRP7 to the microtubule organizing center and the Golgi apparatus in lymphoblastoid cell lines [41], which suggests that the two genes may have similar or overlapping functions in oocyte and early embryonic development.
RHMs Caused by Mutations in NLRP7 or KHDC3L are Mostly Diploid Biparental
Common sporadic nonrecurrent CHMs are mostly diploid androgenetic. Among them, approximately 80 % are monospermic and the remaining are dispermic. Common sporadic PHMs are mostly triploid dispermic. Deviations from these common genotypes, such as monospermic triploidy, digynic triploidy, triandric tetraploidy, biparental tetraploidy, and biparental diploidy, have also been reported among both complete and partial moles but account for a minority of cases, estimated at about 8 % of common moles [44, 45]. This is not the case, however for molar tissues from patients with NLRP7 or KHDC3L mutations. In patients with two NLRP7 defective alleles, the parental contribution to approximately 81 HM tissues has been reported, and all were found to be diploid biparental with the exception of two, which were found to be triploid dispermic [26] and triploid digynic [46] (Table 1). The same applies to patients with two KHDC3L defective alleles. Among these patients, the parental contributions to nine HM tissues have been determined, and all of them were found to be diploid biparental [23, 41, 47, 48] with the exception of one HM tissue that was found to be triploid digynic [46] (Table 1).
Among patients with a single heterozygous NLRP7 mutation or very rare variants not seen in controls, parental contribution to 15 HM tissues has been reported. Of these, four were found to be diploid biparental [26], seven were found to be diploid androgenetic monospermic [21, 27, 30], and four were found to be triploid dispermic [22] (Table 1). With respect to KHDC3L, no molar tissues from patients with single heterozygous variants have been characterized.
Genomic Imprinting in Diploid Biparental Moles
Altered DNA Methylation at Imprinted Genes in the Conceptions of Patients with KHDC3L or NLRP7 Mutations
Genomic imprinting refers to epigenetic modifications such as DNA methylation, histone modification, or/and chromatin remodeling that lead to the expression of only one of the two parental copies of a gene. The involvement of genomic imprinting in the pathology of hydatidiform moles emerged soon after the demonstration that sporadic complete moles are androgenetic, which made them an important experimental tool in characterizing the expression and/or methylation of imprinted genes [49–51]. Later, the identification of recurrent familial moles that have the same histopathological features as the sporadic androgenetic moles [52] and the finding that these moles are diploid biparental [53] entertained the plausible and interesting idea that the causative gene for recurrent moles would be responsible for setting or maintaining the maternal imprints in the oocytes. To date, four studies have examined the DNA methylation of imprinted genes in a total of eight diploid biparental hydatidiform moles from patients with two defective alleles in KHDC3L or NLRP7 [23, 47, 54, 55]. The first study demonstrated, in one diploid biparental CHM from a patient with two KHDC3L defective alleles, the loss of methylation marks at six of seven analyzed differentially methylated regions (DMR) that are normally maternally methylated, and the gain of methylation marks on one paternally methylated DMR (NESP55) that acquires its methylation at the blastocyst stage (Table 2). In contrast, the methylation at the H19 DMR, which is normally established in the male germ line, was normal. Two additional diploid biparental moles from the same patient were later studied, but unfortunately at different DMRs, and their analysis showed the same trend of abnormal methylation with the exception of one gene, PEG10, which preserved its normal methylation on the maternal allele (Table 2) [23]. Other studies also examined the methylation status of DMRs in moles from patients with two NLRP7 defective alleles and reported abnormal loss and gain of methylation at some of them [23, 54, 55]. In one of these studies, single nucleotide polymorphisms were used to distinguish parental alleles at some imprinted genes and showed that the abnormal methylation, indeed, affected the maternal alleles [54] (Table 2). In conclusion, these data demonstrated the presence of imprinting abnormalities in diploid biparental moles from patients with KHDC3L or NLRP7 mutations and indicated that these abnormalities may have occurred during oogenesis and/or early embryogenesis. Because NLRP7 and KHDC3L proteins do not have DNA binding domains or any domain that is found in DNA methyltransferases, it was not clear whether these methylation defects play a primary causal role in the oocytes leading to the formation of moles or whether they are a secondary consequence of abnormal postzygotic development.
Altered DNA Methylation Beyond Non-Imprinted Genes
To investigate the role of NLRP7 in establishing methylation marks at imprinted genes, Mahadevan et al. recently examined the consequences of NLRP7 knockdown on the DNA methylation of imprinted genes during the differentiation of human embryonic stem cells (hESCs) into trophoblast cells [35•]. However, they did not observe any DNA methylation changes at imprinted DMRs, including those that were previously shown to be abnormally methylated in diploid biparental molar tissues. They explained their findings by the known high degree of epigenetic stability and resistance of hESC lines to perturbations in DNA methylation at imprinted loci [56]. Conversely, they found that NLRP7 knockdown altered the DNA methylation of many non-imprinted CpGs. Another interesting study showed that the DNA methylation of a total 131 imprinted and non-imprinted loci were altered in blood DNA of an individual with multiple anomalies born to a mother with a single heterozygous NLRP7 mutation (A719V) [57]. It would have been interesting in this study to have determined if the mutation in the mother occurred de novo or if it was inherited, and from which of her parents. In addition, it is not clear whether the abnormal child inherited his mother’s mutation. Surprisingly, comparing the abnormally methylated genes from the studies by Mahadevan et al. and Beygo et al. [35•, 57] did not reveal any common gene with altered methylation, which raise questions about the specificity and significance of these findings and their relation to NLRP7 mutations that remain to be clarified in future studies.
Altered Expression of CDKN1C in the Conceptions of Patients with NLRP7 Mutations
In line with the above data, one study demonstrated the underexpression of p57KIP2, the product of the paternally imprinted, maternally expressed gene CDKN1C in the cytotrophoblast and villous stroma of a series of diploid biparental CHMs [58]. p57KIP2 is the protein coded by a cyclin-dependent kinase inhibitor. CDKN1C deficiency in mice leads to altered cellular proliferation and differentiation, resulting in a variety of developmental defects [59]. Although CDKN1C is paternally methylated in the cytotrophoblast and villous stroma of normal first-trimester placenta, its expression has been shown to depend on the maternal methylation of KvDMR1, a CpG island located at the promoter of KCNQ1OT1 believed to control the imprinted expression of CDKN1C during embryonic development [60, 61]. The same is observed in humans, where the loss of maternal methylation marks at KvDMR1 leads to the silencing of CDKN1C in patients with Beckwith-Wiedemann syndrome [62], a pediatric overgrowth disorder in which the placenta share some histopathological features with PHMs.
Despite the complexity of the methylation and imprinting data and the variations between studies and samples, the common findings were the lack of DNA maternal methylation marks at several maternally imprinted, paternally expressed genes and the unspecific/stochastic extension of methylation abnormalities to non-imprinted genes. We believe that further studies are needed to delineate the exact roles of NLRP7 and KHDC3L genes in the DNA methylation of imprinted and non-imprinted genes.
Conclusions
NLRP7 and KHDC3L DNA Testing
Because of the high rate of NLRP7 mutations in patients with RHMs, which seems to vary with populations, patients with at least two HMs (complete or partial) should be first offered NLRP7 DNA testing that is now available in many clinical and research laboratories, including ours. Methods currently in use rely on PCR amplification of the 11 exons of NLRP7 from genomic DNA, followed by direct sequencing of the PCR products in the two directions and the comparison of the sequences with the reference sequence NM_001127255.1. This analysis is highly sensitive in identifying point mutations within the coding region, small deletions and insertions affecting the amplified regions, and DNA changes at the invariant splice sites. However, this method is not reliable to identify deep intronic single-nucleotide changes affecting the splicing of the gene or regulatory sequences, large deletions, insertions, and complex rearrangements.
Patients without NLRP7 mutations should be screened for KHDC3L mutations, which account for up to 14 % of cases who are NLRP7-negative [40•, 41, 63]. Similarly, for KHDC3L, currently methods rely on PCR amplification of its 3 exons from genomic DNA, followed by direct sequencing of the PCR products in the two directions and the comparison of the sequences with the reference sequence NM_001017361. Because of the causal involvement of this gene in a minority of cases, only some of the laboratories that offer NLRP7 testing are currently systematically sequencing KHDC3L for all patients who are NLRP7-negative. The identification of two defective alleles in either gene allows to confirm a genetic defect underlying mole recurrence and to counsel the patients accordingly based on available data in the field.
Prognosis for Future Pregnancies
The goal of patients seeking DNA testing is to ascertain their chances of conceiving healthy babies and their risk for mole recurrence and malignant sequelae. Studies from various groups have shown that the chances of a normal live birth are very low in women with two defective alleles in NLRP7. Among reported patients from our group, only 3 out of 43 (7 %) had normal live births, which accounted for 1.5 % of their pregnancies. No other cases of live births have been reported by other groups in patients with RHMs and NLRP7 or KHDC3L mutations, with the exception of a recently described case of one live birth to a patient with two defective alleles in NLRP7 [35•]. In the few reported patients with two KHDC3L defective alleles, no live birth has been reported.
Based on available data, both genes NLRP7 and KHDC3L are required in the oocytes. Therefore, theoretically, ovum donation is expected to improve patients’ reproductive outcomes. Thus far, few patients with mutations in NLRP7 have tried ovum donation, and three had normal live births ([64•] and Slim et al. in preparation), which provides some hope for patients despite the elevated cost of such procedure and its inaccessibility for many of them.
For patients with RHMs and no mutations in either gene, we believe that moles in these patients are most likely to be of a genetic etiology caused by undetected mutations in either NLRP7 or KHDC3L or by mutations in non-identified genes. Based on our current understanding of moles and their mode of formation, the best help that can be offered to these patients is to review the histopathology of their moles and determine the parental contribution to the molar genomes. Based on the results of this analysis, the patients may be classified into two categories:
-
1.
Patients with at least two diploid biparental moles that fit the histological classification of complete molar pregnancies can be counselled in the same way as patients with two NLRP7 or KHDC3L mutations despite the lack of identified mutations. In patients where the histopathological re-evaluation disagrees with the diagnosis of moles, such cases can be counselled similarly to patients with recurrent spontaneous abortions.
-
2.
Patients with androgenetic or triploid dispermic moles have higher chances of having live births from their own oocytes than patients with recurrent diploid biparental moles. Because androgenetic and triploid dispermic moles are caused by errors that occur either at the time of fertilization or very early in the zygote, these patients may benefit from in vitro fertilization followed by preimplantation genetic screening (PGS) to select for diploid embryos to be transferred to the patients. This option may not completely prevent having additional moles, but may help to maximize the patients’ chances of having normal pregnancies, which should be monitored according to standard prenatal care for women with recurrent reproductive loss. A chart summarizing our suggested approach for DNA testing and genetic counselling of patients with RHMs is provided in Fig. 2.
Fig 2 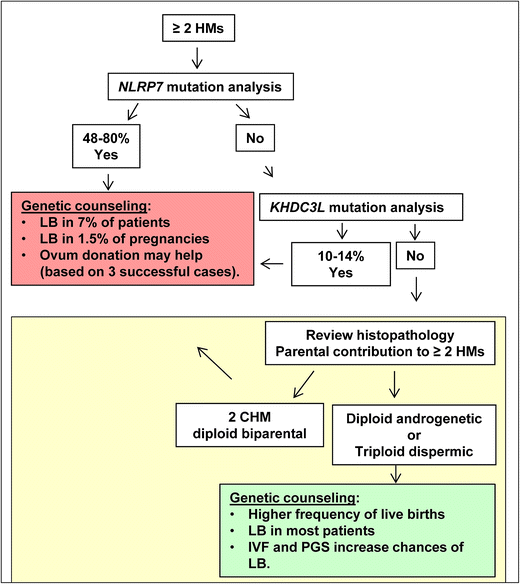
Suggested screening recommendation for DNA testing and genetic counselling of patients with recurrent hydatidiform moles. Patients with at least two HMs should be offered DNA testing first for NLRP7, in which mutations are found in 48–80 % of such patients. Among those with two mutated alleles, up to 7 % may have normal live birth (LB) from their own oocytes in 1.5 % of their pregnancies. To date, three cases of successful ovum donation have been observed in such patients. Patients without NLRP7 mutations should be tested for KHDC3L, in which mutations are found in 10–14 % of such patients. For patients with no mutations in either gene, we propose to re-examine the histopathology of their moles and determine the parental contribution to them. Patients with confirmed complete moles that are diploid biparental can be counselled in the same way as patients with mutations in NLRP7 or KHDC3L. Those with androgenetic or triploid dispermic moles have higher chances of live births from their own oocytes and may be benefit from in vitro fertilization (IVF) and preimplantation genetic screening (PGS) for aneuploidies

Risk for Malignant Degeneration
With respect to the risk for malignant degeneration of moles in patients with mutations in NLRP7 and KHDC3L, we do not have accurate statistics with regard to their risk as compared to patients with sporadic common moles. However, available data on our cases and on those reported by other groups indicate that despite their higher risk for mole recurrence, patients with mutations in either gene are at least not at higher risk for choriocarcinoma. However, future studies are needed to delineate their risk for the less severe neoplastic degeneration and requirement of chemotherapy.
References
Papers of particular interest, published recently, have been highlighted as:• Of importance
Savage P, Williams J, Wong SL, Short D, Casalboni S, Catalano K, et al. The demographics of molar pregnancies in England and Wales from 2000–2009. J Reprod Med. 2010;55(7–8):341–5.
Grimes DA. Epidemiology of gestational trophoblastic disease. Am J Obstet Gynecol. 1984;150(3):309–18.
Bracken MB, Brinton LA, Hayashi K. Epidemiology of hydatidiform mole and choriocarcinoma. Epidemiol Rev. 1984;6:52–75.
Bracken MB. Incidence and aetiology of hydatidiform mole: an epidemiological review. Br J Obstet Gynaecol. 1987;94(12):1123–35.
Berkowitz RS, Im SS, Bernstein MR, Goldstein DP. Gestational trophoblastic disease. Subsequent pregnancy outcome, including repeat molar pregnancy. J Reprod Med. 1998;43(1):81–6.
Kim JH, Park DC, Bae SN, Namkoong SE, Kim SJ. Subsequent reproductive experience after treatment for gestational trophoblastic disease. Gynecol Oncol. 1998;71(1):108–12.
Lan Z, Hongzhao S, Xiuyu Y, Yang X. Pregnancy outcomes of patients who conceived within 1 year after chemotherapy for gestational trophoblastic tumor: a clinical report of 22 patients. Gynecol Oncol. 2001;83(1):146–8.
Van Thiel DH, Ross GT, Lipsett MB. Pregnancies after chemotherapy of trophoblastic neoplasms. Science. 1970;169(952):1326–7.
Rai R, Regan L. Recurrent miscarriage. Lancet. 2006;368(9535):601–11.
Lejeune V. Early recurrent spontaneous abortion: how to take care in 2006? Gynecol Obstet Fertil. 2006;34(10):927–37.
Stephenson M, Kutteh W. Evaluation and management of recurrent early pregnancy loss. Clin Obstet Gynecol. 2007;50(1):132–45. doi:10.1097/GRF.0b013e31802f1c28.
Mack CH, Catherwood AE. The Ascheim-Zondek reaction in hydatidiform moles and malignant chorionepithelioma. Am J Obstet Gynecol. 1930;20:670–8.
Lurain JR, Sand PK, Carson SA, Brewer JI. Pregnancy outcome subsequent to consecutive hydatidiform moles. Am J Obstet Gynecol. 1982;142(8):1060–1.
Sebire NJ, Fisher RA, Foskett M, Rees H, Seckl MJ, Newlands ES. Risk of recurrent hydatidiform mole and subsequent pregnancy outcome following complete or partial hydatidiform molar pregnancy. BJOG: An Int J Obstet Gynaecol. 2003;110(1):22–6. doi:10.1046/j.1471-0528.2003.02388.x.
Yapar EG, Ayhan A, Ergeneli MH. Pregnancy outcome after hydatidiform mole, initial and recurrent. J Reprod Med. 1994;39(4):297–9.
Acosta-Sison H. The chance of malignancy in a repeated hydatidiform mole. Am J Obstet Gynecol. 1959;78:876–7.
Kronfol NM, Iliya FA, Hajj SN. Recurrent hydatidiform mole: a report of five cases with review of the literature. J Med Liban. 1969;22(4):507–20.
Boufettal H, Coullin P, Mahdaoui S, Noun M, Hermas S, Samouh N. Complete hydatiforme mole in Morocco: epidemiological and clinical study. J Gynecol, Obstet Biol Reprod. 2011;40(5):419–29. doi:10.1016/j.jgyn.2011.02.008.
Lorigan PC, Sharma S, Bright N, Coleman RE, Hancock BW. Characteristics of women with recurrent molar pregnancies. Gynecol Oncol. 2000;78(3 Pt 1):288–92. doi:10.1006/gyno.2000.5871.
Murdoch S, Djuric U, Mazhar B, Seoud M, Khan R, Kuick R, et al. Mutations in NALP7 cause recurrent hydatidiform moles and reproductive wastage in humans. Nat Genet. 2006;38(3):300–2.
Qian J, Cheng Q, Murdoch S, Xu C, Jin F, Chebaro W, et al. The genetics of recurrent hydatidiform moles in China: correlations between NLRP7 mutations, molar genotypes, and reproductive outcomes. Mol Hum Reprod. 2011;17(10):612–9.
Slim R, Bagga R, Chebaro W, Srinivasan R, Agarwal N. A strong founder effect for two NLRP7 mutations in the Indian population: an intriguing observation. Clin Genet. 2009;76(3):292–5.
Hayward BE, De Vos M, Talati N, Abdollahi MR, Taylor GR, Meyer E, et al. Genetic and epigenetic analysis of recurrent hydatidiform mole. Hum Mutat. 2009;30(5):E629–39.
Estrada H, Buentello B, Zenteno JC, Fiszman R, Aguinaga M. The p.L750V mutation in the NLRP7 gene is frequent in Mexican patients with recurrent molar pregnancies and is not associated with recurrent pregnancy loss. Prenat Diagn. 2013:1–4. doi:10.1002/pd.4036.
Milhavet F, Cuisset L, Hoffman HM, Slim R, El-Shanti H, Aksentijevich I, et al. The infevers autoinflammatory mutation online registry: update with new genes and functions. Hum Mutat. 2008;29(6):803–8.
Ulker V, Gurkan H, Tozkir H, Karaman V, Ozgur H, Numanoglu C, et al. Novel NLRP7 mutations in familial recurrent hydatidiform mole: are NLRP7 mutations a risk for recurrent reproductive wastage? Eur J Obstet Gynecol Reprod Biol. 2013;170(1):188–92. doi:10.1016/j.ejogrb.2013.06.028.
Deveault C, Qian JH, Chebaro W, Ao A, Gilbert L, Mehio A, et al. NLRP7 mutations in women with diploid androgenetic and triploid moles: a proposed mechanism for mole formation. Hum Mol Genet. 2009;18(5):888–97. doi:10.1093/hmg/ddn418.
Messaed C, Chebaro W, Di Roberto RB, Rittore C, Cheung A, Arseneau J, et al. NLRP7 in the spectrum of reproductive wastage: rare non-synonymous variants confer genetic susceptibility to recurrent reproductive wastage. J Med Genet. 2011;48(8):540–8. doi:10.1136/jmg.2011.089144.
Landolsi H, Rittore C, Philibert L, Hmissa S, Gribaa M, Touitou I, et al. NLRP7 mutation analysis in sporadic hydatidiform moles in Tunisian patients: NLRP7 and sporadic mole. Arch Pathol Lab Med. 2012;136(6):646–51.
Dixon PH, Trongwongsa P, Abu-Hayyah S, Ng SH, Akbar SA, Khawaja NP, et al. Mutations in NLRP7 are associated with diploid biparental hydatidiform moles, but not androgenetic complete moles. J Med Genet. 2012;49(3):206–11. doi:10.1136/jmedgenet-2011-100602.
Manokhina I, Hanna CW, Stephenson MD, McFadden DE, Robinson WP. Maternal NLRP7 and C6orf221 variants are not a common risk factor for androgenetic moles, triploidy and recurrent miscarriage. Mol Hum Reprod. 2013. doi:10.1093/molehr/gat019.
Kinoshita T, Wang Y, Hasegawa M, Imamura R, Suda T. PYPAF3, A PYRIN-containing APAF-1-like protein, is a feedback regulator of caspase-1-dependent interleukin-1{beta} secretion. J Biol Chem. 2005;280(23):21720–5.
• Messaed C, Akoury E, Djuric U, Zeng J, Saleh M, Gilbert L, et al. NLRP7, a nucleotide oligomerization domain-like receptor protein, is required for normal cytokine secretion and co-localizes with Golgi and the microtubule-organizing center. J Biol Chem. 2011;286(50):43313–23. doi:10.1074/jbc.M111.306191. This study demonstrated that ex vivo lipopolysaccharides-stimulated peripheral mononuclear blood cells from patients with NLRP7 mutations secrete lower levels of IL-1β than control cells and suggested that NLRP7 mutations impair cytokine secretion by affecting microtubule structure and consequently intracellular cytokine trafficking.
Khare S, Dorfleutner A, Bryan NB, Yun C, Radian AD, de Almeida L, et al. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity. 2012;36(3):464–76.
• Mahadevan S, Wen S, Wan YW, Peng HH, Otta S, Liu Z, et al. NLRP7 affects trophoblast lineage differentiation, binds to overexpressed YY1 and alters CpG methylation. Hum Mol Genet. 2013. doi:10.1093/hmg/ddt457. This study revealed that NLRP7 knockdown accelerates the differentiation of human embryonic stem cell into trophoblast and alters CpG methylation at non-imprinted genes.
Gerard N, Caillaud M, Martoriati A, Goudet G, Lalmanach AC. The interleukin-1 system and female reproduction. J Endocrinol. 2004;180(2):203–12.
Caillaud M, Duchamp G, Gerard N. In vivo effect of interleukin-1beta and interleukin-1RA on oocyte cytoplasmic maturation, ovulation, and early embryonic development in the mare. Reprod Biol Endocrinol. 2005;3:26.
Zheng H, Fletcher D, Kozak W, Jiang M, Hofmann KJ, Conn CA, et al. Resistance to fever induction and impaired acute-phase response in interleukin-1 beta-deficient mice. Immunity. 1995;3(1):9–19.
Abbondanzo SJ, Cullinan EB, McIntyre K, Labow MA, Stewart CL. Reproduction in mice lacking a functional type 1 IL-1 receptor. Endocrinology. 1996;137(8):3598–601.
• Parry DA, Logan CV, Hayward BE, Shires M, Landolsi H, Diggle C, et al. Mutations causing familial biparental hydatidiform mole implicate c6orf221 as a possible regulator of genomic imprinting in the human oocyte. Am J Hum Genet. 2011;89(3):451–8. doi:10.1016/j.ajhg.2011.08.002. This study identified a second gene, KHDC3L, responsible for RHM that will contribute to the identifcation of new proteins and pathways responsible for this pathology.
Reddy R, Akoury E, Phuong Nguyen NM, Abdul-Rahman OA, Dery C, Gupta N, et al. Report of four new patients with protein-truncating mutations in C6orf221/KHDC3L and colocalization with NLRP7. Eur J Hum Genet. 2013;21(9):957–64. doi:10.1038/ejhg.2012.274.
Pierre A, Gautier M, Callebaut I, Bontoux M, Jeanpierre E, Pontarotti P, et al. Atypical structure and phylogenomic evolution of the new eutherian oocyte- and embryo-expressed KHDC1/DPPA5/ECAT1/OOEP gene family. Genomics. 2007;90(5):583–94. doi:10.1016/j.ygeno.2007.06.003.
Zhang P, Dixon M, Zucchelli M, Hambiliki F, Levkov L, Hovatta O, et al. Expression analysis of the NLRP gene family suggests a role in human preimplantation development. PLoS ONE. 2008;3(7):e2755.
Banet N, Descipio C, Murphy KM, Beierl K, Adams E, Vang R, et al. Characteristics of hydatidiform moles: analysis of a prospective series with p57 immunohistochemistry and molecular genotyping. Mod Pathol. 2013. doi:10.1038/modpathol.2013.143.
Sundvall L, Lund H, Niemann I, Jensen UB, Bolund L, Sunde L. Tetraploidy in hydatidiform moles. Hum Reprod. 2013.
Fallahian M, Sebire NJ, Savage PM, Seckl MJ, Fisher RA. Mutations in NLRP7 and KHDC3L confer a complete hydatidiform mole phenotype on digynic triploid conceptions. Hum Mutat. 2013;34(2):301–8.
Judson H, Hayward BE, Sheridan E, Bonthron DT. A global disorder of imprinting in the human female germ line. Nature. 2002;416(6880):539–42.
Landolsi H, Rittore C, Philibert L, Missaoui N, Hmissa S, Touitou I, et al. Screening for NLRP7 mutations in familial and sporadic recurrent hydatidiform moles: report of 2 Tunisian families. Int J Gynecol Pathol. 2011;30(4):348–53. doi:10.1097/PGP.0b013e31820dc3b0.
Ghazi H, Magewu AN, Gonzales F, Jones PA. Changes in the allelic methylation patterns of c-H-ras-1, insulin and retinoblastoma genes in human development. Dev Suppl. 1990:115–23.
Clarke A. Genetic imprinting in clinical genetics. Dev Suppl. 1990:131–9.
Miyamoto S, Sasaki M, Nishida M, Wake N. Identification of a chromosome carrying a putative tumor suppressor gene in human choriocarcinoma by microcell-mediated chromosome transfer. Hum Cell. 1991;4(1):38–43.
Kircheisen R, Schroeder-Kurth T. Familial hydatidiform mole syndrome and genetic aspects of this disturbed trophoblast development. Geburtshilfe Frauenheilkd. 1991;51(7):569–71.
Sunde L, Vejerslev LO, Jensen MP, Pedersen S, Hertz JM, Bolund L. Genetic analysis of repeated, biparental, diploid, hydatidiform moles. Cancer Genet Cytogenet. 1993;66(1):16–22.
El-Maarri O, Seoud M, Coullin P, Herbiniaux U, Oldenburg J, Rouleau G, et al. Maternal alleles acquiring paternal methylation patterns in biparental complete hydatidiform moles. Hum Mol Genet. 2003;12(12):1405–13.
Kou YC, Shao L, Peng HH, Rosetta R, del Gaudio D, Wagner AF, et al. A recurrent intragenic genomic duplication, other novel mutations in NLRP7 and imprinting defects in recurrent biparental hydatidiform moles. Mol Hum Reprod. 2008;14(1):33–40. doi:10.1093/molehr/gam079.
Rugg-Gunn PJ, Ferguson-Smith AC, Pedersen RA. Status of genomic imprinting in human embryonic stem cells as revealed by a large cohort of independently derived and maintained lines. Hum Mol Genet. 2007;16(Spec No. 2):R243-51. doi:10.1093/hmg/ddm245.
Beygo J, Ammerpohl O, Gritzan D, Heitmann M, Rademacher K, Richter J, et al. Deep bisulfite sequencing of aberrantly methylated Loci in a patient with multiple methylation defects. PLoS One. 2013;8(10):e76953. doi:10.1371/journal.pone.0076953.
Fisher RA, Hodges MD, Rees HC, Sebire NJ, Seckl MJ, Newlands ES, et al. The maternally transcribed gene p57(KIP2) (CDNK1C) is abnormally expressed in both androgenetic and biparental complete hydatidiform moles. Hum Mol Genet. 2002;11(26):3267–72.
Zhang P, Liegeois NJ, Wong C, Finegold M, Hou H, Thompson JC, et al. Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature. 1997;387(6629):151–8.
Fitzpatrick GV, Soloway PD, Higgins MJ. Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat Genet. 2002;32(3):426–31. doi:10.1038/ng988.
Bhogal B, Arnaudo A, Dymkowski A, Best A, Davis TL. Methylation at mouse Cdkn1c is acquired during postimplantation development and functions to maintain imprinted expression. Genomics. 2004;84(6):961–70. doi:10.1016/j.ygeno.2004.08.004.
Diaz-Meyer N, Day CD, Khatod K, Maher ER, Cooper W, Reik W, et al. Silencing of CDKN1C (p57KIP2) is associated with hypomethylation at KvDMR1 in Beckwith-Wiedemann syndrome. J Med Genet. 2003;40(11):797–801.
Zhao W, Muhetaer A, Luo T, Zhou W, Qi C, Chen X, et al. Absence of KHDC3L mutations in Chinese patients with recurrent and sporadic hydatidiform moles. Cancer Genet. 2013. doi:10.1016/j.cancergen.2013.09.003.
• Fisher RA, Lavery SA, Carby A, Abu-Hayyeh S, Swingler R, Sebire NJ, et al. What a difference an egg makes. Lancet. 2011;378(9807):1974. doi:10.1016/S0140-6736(11)61751-0. This report established for the first time that oocyte donation helps patients with NLRP7 mutations to achieve normal pregnancies, indicating that the primary defect caused by NLRP7 mutations occurs in the oocyte.
Helwani MN, Seoud M, Zahed L, Zaatari G, Khalil A, Slim R. A familial case of recurrent hydatidiform molar pregnancies with biparental genomic contribution. Hum Genet. 1999;105(1–2):112–5.
Sensi A, Gualandi F, Pittalis MC, Calabrese O, Falciano F, Maestri I, et al. Mole maker phenotype: possible narrowing of the candidate region. Eur J Hum Genet. 2000;8(8):641–4.
Panichkul PC, Al-Hussaini TK, Sierra R, Kashork CD, Popek EJ, Stockton DW, et al. Recurrent biparental hydatidiform mole: additional evidence for a 1.1-Mb locus in 19q13.4 and candidate gene analysis. J Soc Gynecol Investig. 2005;12(5):376–83.
Zhao J, Moss J, Sebire NJ, Cui QC, Seckl MJ, Xiang Y, et al. Analysis of the chromosomal region 19q13.4 in two Chinese families with recurrent hydatidiform mole. Hum Reprod. 2006;21(2):536–41.
Qian J, Deveault C, Bagga R, Xie X, Slim R. Women heterozygous for NALP7/NLRP7 mutations are at risk for reproductive wastage: report of two novel mutations. Hum Mutat. 2007;28(7):741.
Buyukkurt S, Fisher RA, Vardar MA, Evruke C. Heterogeneity in recurrent complete hydatidiform mole: presentation of two new Turkish families with different genetic characteristics. Placenta. 2010;31(11):1023–5. doi:10.1016/j.placenta.2010.09.003.
Sebire NJ, Savage PM, Seckl MJ, Fisher RA. Histopathological features of biparental complete hydatidiform moles in women with NLRP7 mutations. Placenta. 2013;34(1):50–6.
Brown L, Mount S, Reddy R, Slim R, Wong C, Jobanputra V, et al. Recurrent pregnancy loss in a woman with NLRP7 mutation: not all molar pregnancies can be easily classified as either “partial” or “complete” hydatidiform moles. Int J Gynecol Pathol. 2013;32(4):399–405. doi:10.1097/PGP.0b013e31826cbf6a.
Slim R, Ao A, Surti U, Zhang L, Hoffner L, Arseneau J, et al. Recurrent triploid and dispermic conceptions in patients with NLRP7 mutations. Placenta. 2011;32(5):409–12.
Acknowledgment
Ngoc Minh Phuong Nguyen was supported by the Réseau Québécois en Reproduction, the Alexander McFee McGill Faculty of Medicine Fellowships, and the Canadian Institute for Health Research for RS (MOP-102469).
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Ngoc Minh Phuong Nguyen and Rima Slim declare that they have no conflict of interest
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Nguyen, N.M.P., Slim, R. Genetics and Epigenetics of Recurrent Hydatidiform Moles: Basic Science and Genetic Counselling. Curr Obstet Gynecol Rep 3, 55–64 (2014). https://doi.org/10.1007/s13669-013-0076-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13669-013-0076-1