Article Text

Abstract
Objective Preclinical evidence and early clinical trials have demonstrated the activity of SPL-108, a targeted agent that inhibits CD44 mediated induction of multidrug resistance specifically to paclitaxel and platinum agents. We conducted a phase I, open label, dose escalation study of the safety and tolerability of the combination of SPL-108 with weekly paclitaxel in patients with platinum resistant CD44+ ovarian, primary peritoneal, or fallopian tube cancer.
Methods Patients with platinum resistant histologically proven epithelial ovarian, primary peritoneal, or fallopian tube cancers and measurable disease according to RECIST (Response Evaluation Criteria in Solid Tumours) version 1.1 were selected. Tumors were tested for CD44 expression for eligibility, defined as strong (+++) or moderate (++) staining in ≥20% of the tumor tissue or diffuse + staining. Patients were treated with daily and then twice daily SPL-108 subcutaneous injections and weekly intravenous paclitaxel on days 1, 8, and 15 of a 28 day cycle. Endpoints included safety, determination of maximum tolerated dose, and efficacy. Tumors underwent comprehensive genomic profiling, and cell lines and western blotting were used to study markers of response.
Results We screened 16 patients, and 14 were enrolled based on CD44+ expression. A total of 86% of patients had high grade serous tumors and all had received multiple prior therapies. There were no grade 4–5 toxicities. One patient had grade 3 peripheral sensory neuropathy attributed to paclitaxel and one patient developed presumed colonic perforation attributed to the study drug. No dose reductions or treatment discontinuations were required. All patients tolerated the maximum planned dose; no maximum tolerated dose was reached. Overall response rate was 36%; 5 (36%) patients had partial response and 5 (36%) patients had stable disease.
Conclusions The combination of SPL-108 with weekly paclitaxel was safe and well tolerated. Encouraging antitumor activity was observed, with 72% of patients deriving a clinical benefit.
Trial registration NCT03078400.
- ovarian cancer
- gynecologic surgical procedures
- medical oncology
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
HIGHLIGHTS
SPL-108-paclitaxel is safe and well tolerated for platinum resistant, CD44+ ovarian cancer patients.
Antitumor activity was observed in 36% of patients.
Tumors with TP53 loss of function mutations did not respond to therapy.
What is already known on this topic
The treatment of platinum resistant ovarian cancer is challenging because of short progression free survival rates, making evident that there is a need for new, less toxic and more effective therapies
What this study adds
Our phase I study shows that reversing drug resistance using and the anti-CD44 nanomolecule, SPL-108, in combination with weekly paclitaxel is safe and may improve progression free survival with minimal toxicity in CD44 + platinum refractory ovarian tumors.
How this study might affect research, practice or policy
Future clinical trials will be designed to compare this combination with standard therapies.
Introduction
The global mortality rate for women with ovarian cancer was 207 252 in 2020.1 Survival rates remain low, and 80% of patients who present with advanced disease eventually relapse. While most ovarian cancers are platinum sensitive, tumors eventually become platinum resistant or refractory.2 Ovarian cancer patients with platinum resistant tumors have a poor prognosis because current treatments are usually ineffective. Weekly administration of paclitaxel has been shown to be a useful management approach in women with both platinum and paclitaxel (traditionally administered every 3 weeks) resistant ovarian cancer, with an objective response rate of 20.9–28.9%.3 4 New therapeutic approaches are needed in these patients.
Recurrence in advanced platinum resistant ovarian cancer is linked to a subpopulation of highly invasive tumorigenic cells that are resistant to cytotoxic drugs. These cells are characterized by high expression of CD44, a multifunctional cell surface receptor that modulates cellular processes.5–7 CD44 expression is upregulated in several tumor types, such as breast,8 gastric,9 bladder,10 colon,11 and head and neck cancers.12 In other tumors, there is an observed downregulation of CD44, thus resulting in variable clinical implications in different tumor types.13 CD44 is also implicated in oncogenic phenotypes and is a molecular predictor of survival in ovarian cancer patients.14 Several preclinical studies, including ours, found a link between chemoresistance and CD44 pathways, including MDR1 dependent/P-glycoprotein mediated efflux of chemotherapeutic agents.15 16 We previously showed that CD44 inhibits P-glycoprotein degradation, thus increasing P-glycoprotein mediated drug resistance.17 This resistance may result in selective expansion of invasive cell populations following firstline chemotherapy, leading to recurrence. We demonstrated that targeting CD44 or related signaling pathways, suppresses tumor growth and increases sensitivity to chemotherapeutic agents, such as paclitaxel.18
SPL-108 (Splash Pharmaceuticals Inc, San Diego, California, USA) is an 8 amino acid peptide (Online Supplemental Figure 1) that binds to CD44 and modulates its actions. CD44 is the main ligand of hyaluronic acid. SPL-108 increases binding of CD44 expressing SKOV3 (ovarian cancer) cells to hyaluronan coated plates.19 This is blocked with IM7 (anti-CD44 antibody). Conversely, neither SPL-108 nor IM7 have any effect on binding of CD44 non-expressing A2780 (epithelial ovarian cancer) cells to hyaluronan coated plates. Binding of SPL-108 to CD44 also modulates CD44 mediated intracellular signaling,19 establishing a functional relationship between SPL-108 and CD44 in CD44 expressing cells.
Supplemental material
SPL-108 has antiangiogenic, antimigratory, and anti-invasive properties against cancer cells in vitro.20 SPL-108 inhibits major vessel formation and branching morphogenesis in a model of basic fibroblast growth factor induced angiogenesis in a 7-day-old chick chorioallantoic membrane model.21 SPL-108 has therapeutic activity in in vivo models of melanoma, prostate cancer, glioblastoma multiformae, ovarian, and breast cancer, with mild self-limited adverse events. In a phase Ib clinical trial, daily subcutaneous injection of SPL-108 was well tolerated at all dose levels (150–300 mg daily), without dose limiting toxicity in 16 women with advanced gynecologic malignancies. In this and prior studies, the daily dose of 300 mg resulted in an area under the time–concentration curve similar to that observed in mice at therapeutic doses and was therefore selected as the highest dose for subsequent phase II trials. Additionally, in a phase II trial in 24 patients with recurrent ovarian cancer, treatment with SPL-108, 150 or 300 mg daily, was associated with a statistically significant improved time to disease progression in comparison with placebo.22 For the trial reported here, we chose to start the first cohort with one dose level below the phase 2 dose (P2D) (150 mg, given concurrent paclitaxel) and dose escalate to 300 mg daily (qD) for the second cohort.
Based on the biological rationale that (1) SPL-108 inhibition of CD44 will significantly decrease P-glycoprotein expression, (2) P-glycoprotein over expression is one of the known mechanisms of paclitaxel drug resistance, and (3) SPL-108 in combination with paclitaxel demonstrates activity in ovarian cancer in preclinical studies, we tested SPL-108 and paclitaxel in combination in patients with platinum resistant CD44+ ovarian cancer to assess safety, tolerability, and preliminary estimates of efficacy of this novel treatment approach and examined molecular markers of response.
Methods
Trial Design
This was a phase I, open label clinical study of SPL-108 and paclitaxel using a standard '3+3' dose escalation design, including two different dose levels of eligible patients (Figure 1), followed by expansion cohort. Patients on dose level 1 received SPL-108 150 mg subcutaneously every 24 hours with 80 mg/m2 paclitaxel weekly intravenously on days 1, 8, and 15 of a 28 day cycle. Patients on dose level 2 and the subsequent expansion cohort received SPL-108 150 mg subcutaneously every 12 hours with 80 mg/m2 paclitaxel weekly intravenously with the same schedule.

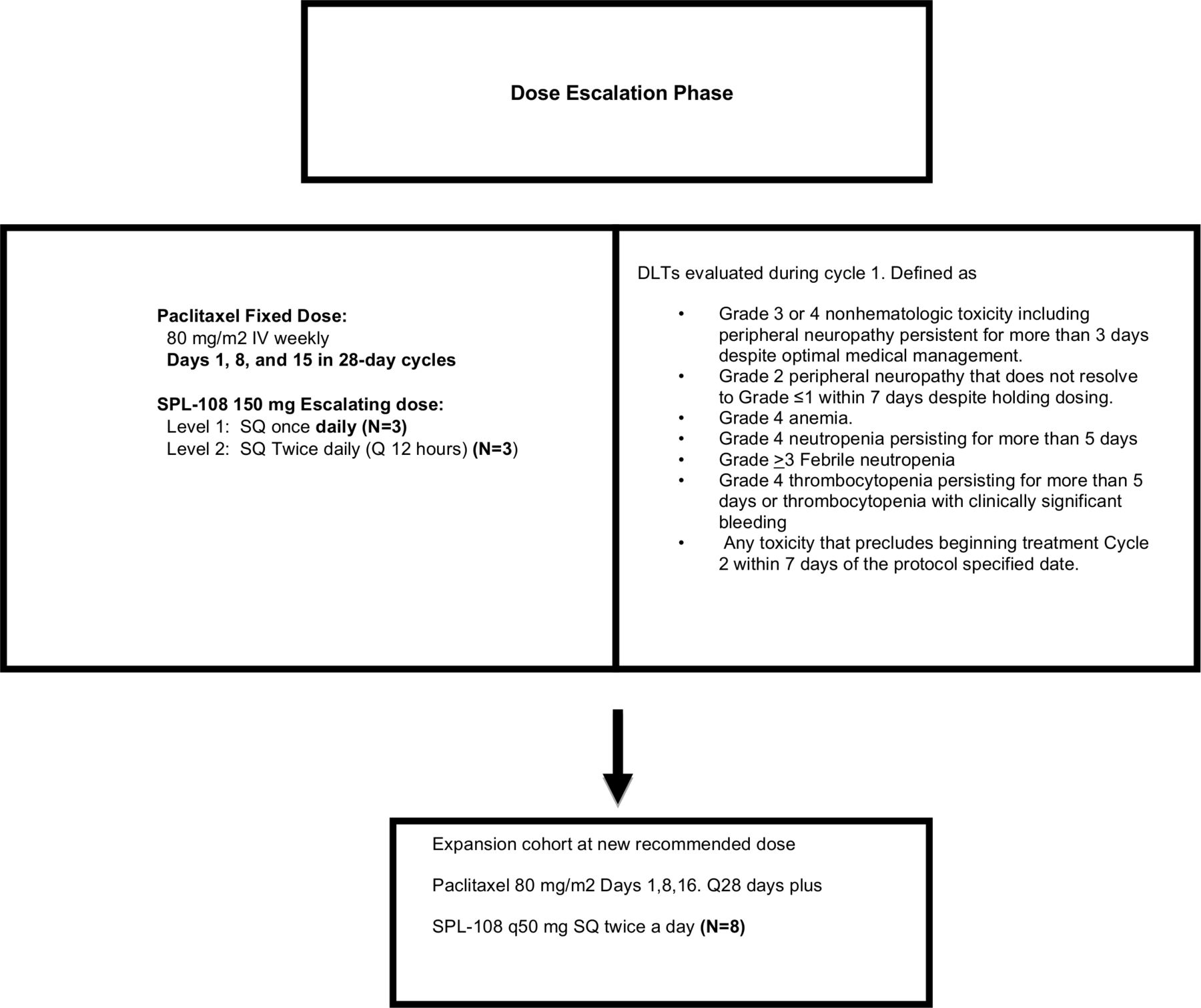{kind=link}
Study design. DLT, dose limiting toxicity; SPL-108, study drug; SQ, subcutaneous.
SPL-108 injections were supplied in prefilled syringes. At the beginning of each cycle, subjects were dispensed one or two kits containing 30 prefilled syringes, depending on the dose level assigned. Study staff educated patients on administration, and each kit contained a unique identifier and a package insert with directions for use. Patients were required to complete a dosing diary, and treatment compliance was monitored at each visit.
Rutgers Cancer Institute of New Jersey, Columbia University Irvings Medical Center (Columbia), and City of Hope Comprehensive Cancer Center participated in the study. Patients were enrolled from Rutgers Cancer Institute of New Jersey and Columbia, and the institutional review boards of these two institutions approved the study. All subjects provided written informed consent to participate in the study, before the first study specific procedure.
Study Population
Patients were enrolled between May 2017 and December 2018. Patients were eligible if they were 18 years of age or older with either histologically or pathologically proven epithelial ovarian cancer, or primary peritoneal or fallopian tube cancer, as reviewed by an expert pathologist (MC) at Rutgers Cancer Institute of New Jersey. Patients were required to complete at least one prior line of platinum based therapy. Platinum resistant or refractory tumors were defined by recurrence of disease within 6 months of completion of platinum based therapy or disease progression during or within 1 month of completing platinum based therapy, respectively. Tumors were tested for CD44 expression by immunohistochemistry, defined as strong (+++) or moderate (++) staining in ≥20% of the tumor tissue or diffuse+ staining, using a Clinical Laboratory Improvement Amendments certified test. Most patients had a study entry biopsy or cytology, but original biopsy was allowed if patient declined a new biopsy.
Primary End Points
The primary endpoint was maximum tolerated dose and safety of SPL-108 subcutaneously in combination with weekly intravenous paclitaxel. Dose limiting toxicities were evaluated during the first cycle of treatment. Secondary endpoints included best response, clinical response rate, and progression free survival. Exploratory endpoints included laboratory correlates examining markers of response.
Sample Size
Sixteen patients were selected. If one patient experienced a dose limiting toxicity, three additional patients were enrolled at that dose level. If no patients experienced a dose limiting toxicity or only one of six patients experienced a dose limiting toxicity, a second cohort (cohort 2) was enrolled at dose level 2 and treated with SPL-108 300 mg daily (150 mg subcutaneously every 12 hours) with concurrent 80 mg/m2 paclitaxel weekly on days 1, 8, and 15 of a 28 day cycle. An expansion cohort of six patients was planned at the highest dose if no maximum tolerated dose was observed.
Statistical Methods
Progression free survival estimates with 95% confidence intervals (CI) were calculated using complementary log–log transformation of progression free survival estimates. Categorical variables are presented in frequency tables. Laboratory correlates were performed in triplicate and the t test used for significance estimates.
Clinical Toxicity Evaluation
All patients who received at least one cycle of protocol therapy were evaluated for toxicity. Adverse events were assessed weekly according to the National Cancer Institute terminology criteria for Adverse Events (CTCAE) version 4.0. A dose limiting toxicity was defined as an adverse event that met one of the following criteria: (1) treatment related ≥grade 3 non-hematologic toxicity, excluding alopecia, hypersensitivity reactions, and injection site reactions; (2) grade 4 neutropenia for at least 7 days, febrile neutropenia, and thrombocytopenia accompanied by bleeding; or (3) ≥grade 3 hematologic toxicity, excluding anemia and lymphocytopenia. Maximum tolerated dose was defined as the dose below the dose at which at least two of six patients experienced dose limiting toxicities.
Clinical Response Evaluation
Patients were evaluated for response of measurable disease using computerized tomography (CT) imaging of the chest/abdomen/pelvis at baseline and after every two cycles of protocol therapy. Progression free survival was defined as the date of registration until disease progression or death, whichever came first (censored by the date of last contact prior to data analysis).
Laboratory Correlates
Cell Lines
The results of these correlates are presented in conjunction with this trial report to clarify the possible mechanism of action of this new drug. This will also aid in the design of the next phase II trial. PEO1 cells (BRCA2 mutated, CD44+ ovarian cancer cell line; Sigma; No 10032308-1VL), SKOV3 cells (ATCC; HTB-77), and OVCAR8 cells (BRCA wildtype, CD44+, human ovarian cancer cell line; gift from Dr Edward Wang, City of Hope National Medical Center) were used. Poly (ADP-ribose) polymerase (PARP) inhibitor resistant OVCAR8 cells were generated by continuous incremental olaparib administration.23
Western Blots
Ovarian cancer cells were lysed on ice for 30 min in a radioimmunoprecipitation assay lysis buffer (No 5872, Cell Signaling Technology). Protein concentrations were measured using the Bio-Rad protein assay (Bio-Rad, Germany) and subjected to 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis, transferred to a polyvinylidene difluoride membrane with subsequent immunodetection using anti-MDR1/ABCB1, rabbit monoclonal (Cell Signaling Technology), anti-GAPDH, mouse monoclonal, and anti-VEGF, mouse monoclonal (both from Santa Cruz Biotechnology). All antibodies were diluted in Tween 20 buffer containing 5% bovine serum albumin and detected by enhanced chemiluminescence SuperSignal West Femto substrate (No 34095, Thermo Fischer Scientific) using ChemiDoc MP Imaging System (Bio-Rad). Results of three independent experiments were quantified using ImageJ (NIH) software.
Results
Patient Characteristics
Fourteen of 16 patients had CD44+ tumors and were enrolled in the study. Median age was 60.5 years (45–77). Most patients had high grade serous carcinoma histology (n=12, 86%) and platinum resistant disease (n=12, 86%). Ten patients had received ≤3 prior chemotherapy regimens; four patients had received 4–5 prior regimens. Seven patients (50%) had received prior PARP inhibitor therapy and six (43%) had received prior bevacizumab (Table 1).
Baseline patient characteristics
Safety Profile
Eighty-two treatment cycles were administered with a median of four cycles (range 2–14). Eight patients (57%) received ≥4 cycles. There were no grade 4–5 toxicities. One patient experienced grade 3 peripheral sensory neuropathy attributed to paclitaxel. An additional patient developed a presumed colonic perforation attributed to the study drug, described below (Table 2). There were no injection site reactions or dermatological adverse events and no treatment discontinuations or treatment related deaths. No dose reductions were required. All patients tolerated the maximum planned dose well; no maximum tolerated dose was reached. Six patients completed dose escalation. Eight additional patients completed the expansion phase.
All adverse events, according to National Cancer Institute terminology criteria for Adverse Events (CTCAE) version 4.0
The most salient toxicity, observed in one patient, was grade 3 possible colonic perforation from colon to tumor, not evident on imaging studies or the final pathology. Findings at surgery included dense fibrosis at sites of previous disease with microscopic disease at the abdominal wall mass.
Clinical Response
Promising clinical benefit was observed. Median progression free survival was 110 days (95% CI 56 to 196) (Table 3; Table 4; Online Supplemental Figure 2). Overall response rate was 36%; five patients had a partial response with a median duration of 10 months. Five patients (36%) had stable disease and four (29%) had progression of disease (Table 3). All patients with partial response as best response had not been treated with a PARP inhibitor. In contrast, three of four patients who had progression of disease as best response received prior PARP inhibitors as their last therapy prior to the trial. There was a correlation between CA-125 and clinical impression of reduced ascites, however; none met CA-125 criteria of response. Seven patients had ascites at the beginning of therapy; none had progression of ascites and one had complete ascites resolution (Online Supplemental Table 1). This correlates with our preclinical data, which showed that SPL-108 had antiangiogenic effects and decreased vascular endothelial growth factor expression in an ovarian cancer cell line (Online Supplemental Figure 5).
Tumor response and TP53 alterations or expression
Progression free survival estimates with 95% CI (using complementary log–log transformation of progression free survival estimates)
Molecular Patterns of Response
Eleven patients (79%) had comprehensive genomic profiling using hybrid capture technology. All tumors had low or intermediate mutational burden. No tumors had increased mutation burden or microsatellite instability. One tumor had an MLH1 truncation, but the tumor was microsatellite stable. One patient had a germline BRCA1 mutation, while three patients had somatic BRCA1 mutation.
All four patients with progression of disease had TP53 alterations with loss of function: three nonsense TP53 truncation alterations and one known loss of function. The three tumors that had TP53 truncations showed complete absence of p53 by standard immunohistochemistry, while the tumor with loss of function, but no truncation, had mutant p53 overexpression. Tumors with either a partial response or stable disease had TP53wt (one) and missense gain of function alterations. One tumor that responded with a partial response had a truncation at the end of the molecule, and p53 was likely still functional. (Table 3, Online Supplemental Figure 3).
Correlative Studies
The effect of SPL-108 on ABCB1 expression was tested using western blot. SPL-108 reduced total levels of ABCB1 in both BRCA wild type (OVCAR8 and SKOV3) and BRCA2 mutated (PEO1) cells, indicating that SPL-108 in vitro properties are not cell type specific or dependent on BRCA expression. Furthermore, SPL-108 is affecting ABCB1 expression, a known downstream target of CD44 (Online Supplemental Figure 4).
We hypothesized that SPL-108 had antiangiogenesis properties based on our observations of clinical response of ascites. We tested OVCAR8 cells (Online Supplemental Figure 5) to determine the effect of SPL-108 on vascular endothelial growth factor as a possible mechanism for the finding. After treatment with SPL-108, both ABCB1 and vascular endothelial growth factor expression were decreased by 50%. Given that patients who had PARP inhibitors as the last therapy prior to the trial did not respond to the trial therapy, we tested a PARP inhibitor resistant cell line derived from the parent cell line OVCAR8 that had both high P-glycoprotein (ABCB1) and CD44 expression. Western blots showed that treatment with SPL-108 decreased ABCB1 expression in parental PARP inhibitor sensitive ovarian cancer cell lines by 50% compared with PARP inhibitor resistant cells (Online Supplemental Figure 5). These results will inform the inclusion criteria of future clinical trials.
Discussion
Summary of Main Results
This phase I trial demonstrated that a combination of SPL-108 and paclitaxel is safe and well tolerated in patients with platinum resistant ovarian cancer. There were only three grade 3 toxicities and no grade 4–5 toxicities. Grade 1–2 increased transaminases and development of a possible bowel microperforation (grade 3) were adjudicated to treatment drug. We consider the bowel microperforation as an experimental drug induced complication, although there was no imaging or pathologic proof of a fistula being present because that was the admission diagnosis and management. While the maximum tolerated dose was not reached in the present trial, based on reported clinical activity and safety results, we recommend using SPL-108 150 mg subcutaneously twice daily for future phase II studies.
Five patients (35%) showed a response that persisted for 6–12 months. All patients had tumors with CD44 expression; however, the location of CD44 expression (stroma, nucleus, or epithelial membrane) did not correlate with response. Interestingly, patients who had progression of disease had tumors with p53 loss of function; three had nonsense truncations of TP53 and p53 null mutations. Additionally, patients with partial responses had never received PARP inhibitor therapy. Our preclinical data suggest that prior PARP inhibitor exposure may not allow SPL-108 to optimally decrease CD44 or vascular endothelial growth factor expression.
We showed that SPL-108 decreases P-glycoprotein (ABCB1) expression, which supports SPL-108 reaching the CD44 target in vitro. Decreased expression of the ABCB1 transporter provided the rationale for using paclitaxel (an ABCB1 substrate) on patients who were once clinically resistant to the combination of carboplatin/paclitaxel. Although SPL-108 may reverse ABCB1 drug resistance to paclitaxel, it may not be enough to counteract drug resistance developed after PARP inhibitor therapy.
Results in the Context of Published Literature
P53 is a tumor suppressor protein commonly mutated in epithelial ovarian cancer. Weinberg’s group showed that wild type p53 inhibits CD44 expression by binding to a non-canonical p53 binding sequence in the CD44 promoter and p53 absence results in 'de-repressed CD44 expression'.24 In our trial, patients without tumor p53 expression or with TP53 loss of function did not respond to the study regimen. We hypothesize that lack of p53 may lead to CD44 overexpression in vivo as it has already been shown in vitro, offsetting the effect of the SPL-108 dose used and thereby limiting its efficacy. Future trials with SPL-108 will exclude tumors with p53 loss of function. A clinical trial using SPL-108 for the treatment of symptomatic ascites is presently planned. The recommended dose of SPL-108 to be used in future phase II clinical trials will be 150 mg twice a day.
Strengths and Weaknesses
Although the clinical benefit is evident, there was no power to evaluate the efficacy of this drug combination, which is an inherent limitation to all phase I trials. Furthermore, the limited number of patients does not allow for correlation between molecular patterns of response and histologic subtypes, or to make permanent conclusions about the efficacy of the drug regarding TP53 status or prior exposure to PARP inhibitors. A trial comparing the effectiveness of bevacizumab with weekly paclitaxel, pegylated liposomal doxorubicin, or topotecan to chemotherapy alone in recurrent platinum resistant ovarian cancer (AURELIA), reported a median progression free survival of 3.4 months in the chemotherapy alone arm compared with 6.7 months in the chemotherapy plus bevacizumab arm.
Implications for Practice and Future Research
Our results revealed that when removing patients that had TP53 loss of function from the analysis, the median progression free survival was 6 months, similar to the AURELIA trial. We plan to compare a combination of SPL-108 and weekly paclitaxel versus bevacizumab with weekly paclitaxel in future studies.25
Conclusion
SPL-108 was safe when given in combination with weekly paclitaxel. Furthermore, the combination of SPL-108 and paclitaxel may have similar median progression free survival as a combination of paclitaxel with bevacizumab, but with less severe adverse events. Future phase II trials will be conducted to compare these two treatment combinations.
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by Rutgers institutional review board (2020000290, protocol No 051607). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors thank Nicola Welch, PhD, CMPP (Whipbird Communications) for assistance with writing and editing the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Correction notice This article has been corrected since it was first published. The open access licence has been updated to CC BY.
Contributors LR-R designed and executed the study, supervised the study, and drafted the manuscript. EG participated in clinical trial accrual and manuscript preparation. DN and MF participated in the trial concept and design. JH, ABM, AL, MS, RS, NC, AIT, and JDW participated in clinical trial accrual. MC was involved in pathology analysis. RV was a clinical research manager. JP was involved in biostatistics. HY, AM, and AK were involved in laboratory correlates. All authors approved the final manuscript. LRR acts as guarantor.
Funding The study was funded by Markel Friendman Accelerator Funds and the drug supplied by Splash Pharmaceuticals, respectively.
Competing interests DN and MF have ownership options in Splash Pharmaceuticals.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.