Article Text

Abstract
Background The role of adjuvant treatment in the intermediate-risk group of patients with early-stage cervical cancer is controversial and is supported by a single randomized Gynecologic Oncology Group (GOG) 92 study performed more than 20 years ago. Recent retrospective studies have shown excellent local control in this group of patients after radical surgery with no additional adjuvant treatment.
Primary Objective To evaluate if adjuvant (chemo)radiation is associated with a survival benefit after radical surgery in patients with intermediate-risk cervical cancer.
Study Hypothesis Radical surgery alone is non-inferior to the combined treatment of radical surgery followed by adjuvant (chemo)radiation in disease-free survival in patients with intermediate-risk cervical cancer.
Trial Design This is a phase III, international, multicenter, randomized, non-inferiority trial in which patients with intermediate-risk cervical cancer will be randomized 1:1 into arm A, with no additional treatment after radical surgery, and arm B, receiving adjuvant external beam radiotherapy±brachytherapy ± concomitant chemotherapy. Patient data will be collected over 3 years post-randomization of the last enrolled patient for primary endpoint analysis or for 6 years for the overall survival analysis.
Major Inclusion/Exclusion Criteria Patients with intermediate-risk early-stage cervical cancer (IB1–IIA), defined as lymph node-negative patients with a combination of negative prognostic factors (tumor size >4 cm; tumor size >2 cm and lymphovascular space invasion; deep stromal invasion >2/3; or tumor-free distance <3 mm) with squamous cell carcinoma or human papillomavirus (HPV)-related adenocarcinoma, are eligible for the trial.
Primary Endpoint Disease-free survival defined as time from randomization to recurrence diagnosis.
Sample Size 514 patients from up to 90 sites will be randomized.
Estimated Dates for Completing Accrual and Presenting Results It is estimated that the accrual will be completed by 2027 (with 3 additional years of follow-up) and primary endpoint results will be published by 2031. Estimated trial completion is by 2034.
Trial Registration NCT04989647.
- Cervical Cancer
- Surgical Oncology
- Radiotherapy
Data availability statement
Data are available upon request.
Statistics from Altmetric.com
Introduction
The intermediate-risk group of patients with early-stage cervical cancer is characterized by negative pelvic lymph nodes and a combination of tumor-related prognostic risk factors such as tumor size ≥2 cm, presence of lymphovascular space invasion, deep stromal invasion, or small tumor-free distance.1 2 The role of adjuvant treatment in these patients remains controversial, and international guidelines currently consider both radical surgery alone or followed by adjuvant (chemo)radiation the standard of care in the treatment of these patients.3 4
The evidence for the efficacy of adjuvant radiotherapy comes from a single randomized Gynecologic Oncology Group (GOG) 92 study of whole pelvic radiotherapy or no further treatment following radical hysterectomy and pelvic lymphadenectomy,2 in which adjuvant radiotherapy was associated with a 47% reduction in the risk of recurrence. Extended follow-up data published in 2006 confirmed the improvement in progression-free survival in the adjuvant radiotherapy group.5 However, the GOG 92 trial suffered from many limitations. The study reflects the diagnostic and treatment standards of two decades ago: tumor size was estimated by visual inspection and palpation, and surgery as the main treatment modality was not standardized by the protocol. The prevalence of risk factors was not balanced in the two arms, with a higher prevalence of lymphovascular space invasion and more patients with tumors >4 cm in the arm receiving radical surgery only. The outcome of the surgery-only arm was very poor by current expectations. The total recurrence rate reached 28%, with 67% pelvic recurrences.
In contrast, a recent retrospective study has compared the outcome of intermediate-risk patients after two treatment strategies in three institutions.6 In one institution adjuvant treatment was not administered, irrespective of tumor size, while the remaining two institutions followed the traditional ‘GOG criteria’ and referred all intermediate-risk patients to adjuvant treatment. Only two of the 127 patients had recurrence in the pelvis after tailored radical surgery in the group without any adjuvant treatment, with a median follow-up of 6 years. None of the oncological outcome variables, such as total relapse rate, overall survival, or disease-specific survival, were different from the control group of patients who had received adjuvant radiotherapy. The local control of the disease in both groups was superior to the outcome of the original GOG trial (pelvic recurrence rate in surgery-only arms 2% vs 19% and, in combined treatment arms, 0% vs 13%). An increasing number of recent retrospective studies have supported these findings and reported no significant benefit of adjuvant treatment in intermediate-risk patients.7–10
The reasons for a better outcome in recent studies are probably multifactorial. More precise pre-operative staging using modern imaging and improved standards of pathologic assessment of both the cervix and lymph nodes could identify extra-uterine disease (parametria and lymph nodes) with better precision, excluding such cases from the intermediate-risk group. In the retrospective study mentioned previously,6 sentinel lymph node biopsy was performed in 80% of patients in the surgery-only group. Micrometastases were detected by pathological ultrastaging in 33 patients; these patients received adjuvant radiotherapy and were excluded as node-positive cases. Consequently, the population, although defined by the same inclusion criteria, was more precisely selected. In addition, the technique of parametrectomy and lymph node dissection has been better standardized, so a higher quality of surgery can contribute to a better outcome in this specific patient sub-group with a higher risk of parametrial and pelvic lymph node involvement.
With regard to these advances in pre-operative staging and surgical techniques, the CERVANTES trial is designed and powered to determine whether radical surgery alone is non-inferior to the combined treatment of radical surgery followed by adjuvant (chemo)radiation in disease-free survival of patients with intermediate-risk cervical cancer.
Methods
Trial Design
CERVANTES is a prospective, international, multicenter, randomized, non-inferiority phase III trial aiming to evaluate if adjuvant (chemo)radiation is associated with disease-free survival benefit after radical surgery in patients with intermediate-risk cervical cancer. The trial is led and funded by the Central and Eastern European Gynecologic Oncology Group (CEEGOG CX-05) with international participation. It is being performed in collaboration with the European Network of Gynecologic Oncology Trials (ENGOT-cx16). Ethics approval was obtained from the Ethics Committee of the General University Hospital in Prague (66/20 S-MEK). The number of international sites is expected to evolve progressively, with up to 90 sites planned.
Participants with intermediate-risk cervical cancer will be randomized after the radical surgery to no further treatment (arm A) or to adjuvant treatment (arm B). Figure 1 shows the trial schema.
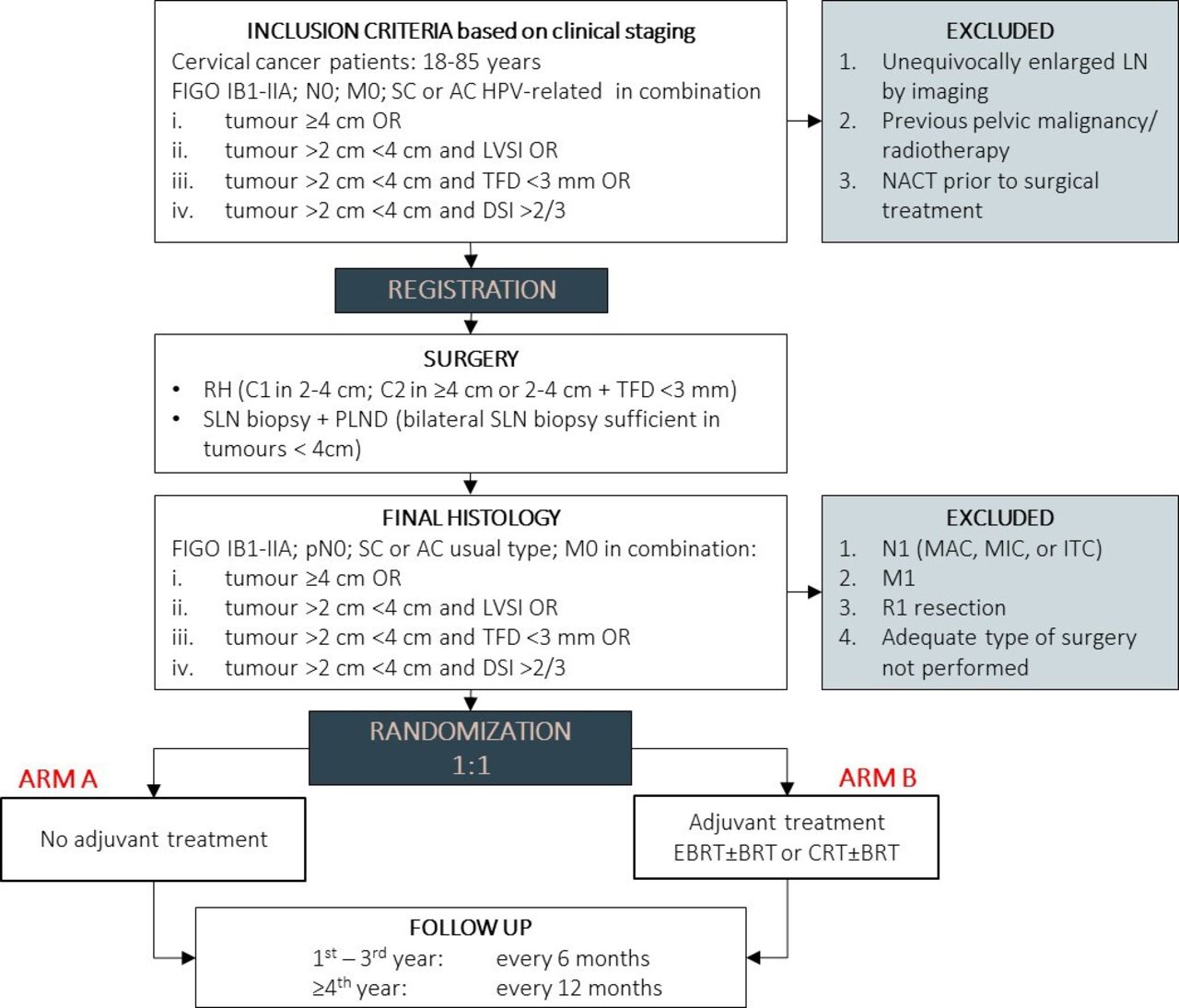
{kind=link}
CERVANTES trial design. AC, adenocarcinoma; BRT, brachytherapy; CRT, chemoradiotherapy; DSI, deep stromal invasion; LVSI, lymphovascular space invasion; EBRT, external beam radiotherapy; M0, no distant metastasis; M1, cancer has spread to other parts of the body; MAC, macrometastasis; MIC, micrometastasis; N0, no lymph node involvement; N1, lymph node involvement; NACT, neoadjuvant chemotherapy; PLND, pelvic lymphadenectomy; R1, positive surgical margins; RH, radical hysterectomy; SC, squamous cell cancer; SLN, sentinel lymph node; TFD, tumor-free distance.
Surgical Treatment
Patients will be registered to the trial before surgery, which will be composed of lymph node staging and radical hysterectomy. Open surgery (laparotomy) is the standard surgical approach. Minimally invasive surgery is acceptable but only with the approval of the Trial Management Committee and only at sites with an extensive experience with such an approach in this cohort of patients. The type of radical hysterectomy should correspond to the prognostic risk factors (based on pre-surgical clinical staging: tumor size, tumor-free distance, and depth of stromal invasion determined by imaging; lymphovascular space invasion determined by biopsy). Type C1 should be performed in tumors <4 cm or in tumors with tumor-free distance >3 mm. If those conditions are not met, type C2 should be performed. Lymph node staging will be composed of sentinel lymph node biopsy or systematic pelvic lymphadenectomy. An attempt at sentinel biopsy is mandatory in all patients and systematic lymphadenectomy can be avoided in patients with tumors <4 cm if sentinel lymph nodes are detected bilaterally.
Adjuvant Treatment
Patients randomized to arm B will receive adjuvant treatment composed of either pelvic radiotherapy (external beam radiotherapy±brachytherapy) or concomitant chemoradiotherapy (external beam radiotherapy±brachytherapy + chemotherapy). The intended strategy of adjuvant treatment will be given at the time of randomization. Adjuvant radiotherapy is standardized in terms of technology, target volumes, dose prescription, and organs at risk. Concomitant chemotherapy, cisplatin or carboplatin-based, will be administered according to the recommended dosage.
Follow-up Procedures
All trial participants, irrespective of treatment group, will be followed until the trial is terminated, which is planned 3 years after the randomization of the last enrolled patient or 6 years in case of a positive outcome in the primary endpoint analysis when the study will continue to collect survival data. In the first 3 years post-randomization, patients will be followed at 6-month intervals for disease status, treatment morbidity, and quality of life. From year 4 onwards, patients will be followed at 12-month intervals for disease status and treatment morbidity.
Participants
Site Selection and Quality Assurance
Both the quality of surgery and the quality of radiotherapy will be assured by the Trial Quality Assurance Committee. The candidate sites will be evaluated for experience and standards of the trial techniques and for access to necessary technical equipment. During the trial, quality assurance at all sites will be continuously monitored for compliance with the protocol procedures and standards.
Patients
Participants registered to the trial must be deemed suitable and fit for radical surgery followed by adjuvant radiotherapy and meet the pre-operative inclusion criteria based on clinical staging (biopsy, expert ultrasound or MRI: imaging, physical examination): (1) International Federation of Gynecology and Obstetrics (FIGO) stage IB1–IIA; (2) squamous cell cancer or human papillomavirus (HPV)-related adenocarcinoma; (3) age 18–85 years; (4) presence of intermediate risk factors: tumor ≥4 cm; tumor >2 cm <4 cm and lymphovascular space invasion or tumor-free distance <3 mm or deep stromal invasion (>2/3) (tumor size, tumor-free distance, and depth of stromal invasion determined by imaging; lymphovascular space invasion determined by biopsy). Patients with unequivocally positive pelvic lymph nodes by imaging or those with previous pelvic malignancy/radiotherapy will be excluded.
Based on the surgical and pathological results, the following groups of patients will be excluded from the trial before randomization: (1) positive lymph nodes (any type of metastasis: isolated tumor cells, micrometastasis, macrometastasis); (2) positive surgical margins; (3) intra-operative findings of extra-uterine spread or upstaging based on final pathology; (4) adequate type of surgery not performed (radical hysterectomy type, adequate lymph node staging); (5) adenosquamous cancer or adenocarcinoma unusual type or other rare tumor types.
Endpoints
The primary endpoint of the study is disease-free survival calculated from the day of randomization until diagnosis of recurrence. In case of a positive outcome of the primary endpoint, the study will continue to collect survival data for the key secondary endpoint, which is overall survival. Other secondary endpoints include evaluation of pelvic disease-free survival, health-related quality of life, and treatment-related adverse events.
Sample Size
A total of 514 eligible patients (including 10% expected drop-out) will be randomized. The sample size for primary endpoint analysis was estimated based on assumptions of the 2-year disease-free survival which is expected to be 85% in arm B, and the maximal tolerated margin for non-inferiority in the 2-year disease-free survival of 5%. To achieve a power of 80% for the hypothesis testing by the Cox proportional hazards model, the number of patients needed for the final analysis is 468. The secondary endpoint of overall survival will be tested using 95% confidence intervals (CI) for the hazard ratio (HR). The sample size of the total 468 evaluable patients results in a power higher than 80% for this secondary testing, assuming 3-year overall survival of 92% in arm B and the maximal tolerated margin of non-inferiority of 5%.
To ensure the safety of the patients during the trial, the Independent Data Monitoring Committee will be responsible for periodic reviews of the data, number of events in each of the trial arms, and occurrence of (serious) adverse events.
Randomization
Patients with pathologically confirmed inclusion criteria will be randomized in a 1:1 ratio into two arms: (1) patients in arm A will not receive any further treatment; (2) patients in arm B will receive adjuvant treatment composed of external beam radiotherapy±brachytherapy ± concomitant chemotherapy. Randomization will be conducted electronically in the CERVANTES electronic Information System developed on the REDCap platform according to a computer-generated randomization list. The patients will be stratified by tumor size (<4 cm versus ≥4 cm) and intended strategy of adjuvant treatment: either external beam radiotherapy ± brachytherapy or concomitant chemoradiotherapy ± brachytherapy.
Statistical Methods
CONSORT guidelines will be followed for the reporting of the trial data. Analysis will be performed according to the intention-to-treat principle.
The primary endpoint will be estimated via the Kaplan–Meier method. The analysis will be evaluated by the HR estimate and its 95% CI using the Cox proportional hazards model comparing two treatment arms. The overall survival will be described using the same methods as those used for the primary endpoint. The non-inferiority margin for both endpoints is set to 5% at a significance level of 0.05 (using 95% CI of HR).
Discussion
Current international guidelines consider both types of management—radical surgery alone and radical surgery followed by adjuvant (chemo)radiation—as the standard of care in the treatment of intermediate-risk cervical cancer.3 4 The available data in the literature are also inconclusive with regard to the inferiority of either approach.6–12
According to the European guidelines, radical surgery should not be performed if an indication for adjuvant radiotherapy is known before the surgery.4 Since tumor size and depth of stromal invasion can be accurately assessed by pre-operative imaging, the majority of cases with intermediate-risk prognostic factors are identified before surgery; these patients can be referred for primary chemoradiation and spared the combined treatment. The main motive for one main treatment modality is the avoidance of additional morbidity associated with the combination of radical surgery and pelvic radiotherapy. Adjuvant radiotherapy increases the risk and worsens the manifestation of certain surgical complications such as urinary fistula and lower limb lymphedema.5 Furthermore, surgery and radiotherapy are associated with a different spectrum of complications, and patients who receive combined treatment have a higher risk of bladder and bowel dysfunction, sexual dysfunction, psychological consequences, and severe impairment of quality of life.13
As stated previously, evidence for adjuvant treatment administration in patients with intermediate-risk cervical cancer comes from a single prospective study. There are, however, several important aspects which distinguish the CERVANTES trial from the original GOG 92 study: (1) patients are registered in the study before surgery, which is part of the protocol with the standardization of surgical performance; (2) a quality assurance program is in place for both surgery and adjuvant treatment; (3) sentinel lymph node biopsy is mandatory (at least an attempt); (4) pathological assessment of the uterus, sentinel and pelvic lymph nodes is standardized by the protocol; (5) an imaging test is mandatory for clinical staging before surgery.
Several aspects of the trial may cause debate or raise questions. (1) Surgical approach: laparotomy is the recommended approach. Minimally invasive surgery requires approval from the Trial Management Committee and it is assumed that it will be accepted only very exceptionally at centers that can demonstrate a high level of experience with endoscopic surgery in these patients, as well as a good patient outcome based on their own audit. The authors are well aware of the results of the LACC study14; however, they consider it less risky to perform a quality procedure with an approach with which the surgeon has extensive experience than to use a surgical approach with which the surgeon is not comfortable only in the patients included in the trial. (2) Type of radical hysterectomy: there is currently no prospective evidence for the recommendation to perform type C2 radical hysterectomy (or another adequate type) in patients with tumors of larger size or deep invasion. However, some retrospective studies have shown a survival benefit of more extensive parametrectomy in larger tumors.7 Assuming that the main reason for parametrectomy in intermediate-risk patients is to remove clinically occult metastases in the lymphatic channels and parametrial lymph nodes, the procedure should aim at complete parametrial resection. (3) Quality assurance: undoubtedly, the quality of treatment, especially the quality of surgical performance, is crucial for the outcome of the study. Therefore, a quality assurance committee was established with the goal to review selected parameters within the selection of sites and during the trial conduct. (4) Lymph node staging: pelvic lymphadenectomy is a mandatory part of the surgical procedure, but bilateral sentinel lymph node detection is sufficient in tumors up to 4 cm. We thus reflect the current clinical practice of many departments which stopped performing systematic lymphadenectomy routinely. The lack of systematic node removal is balanced by the mandatory processing of all sentinel lymph nodes by an ultrastaging protocol which increases the accuracy of surgical staging. (5) Intermediate-risk group diagnostic criteria: compared with the original GOG criteria, a tumor-free distance is a new negative prognostic parameter, which was shown to be better and easier to assess by imaging than the depth of stromal invasion.1 15 (6) Type of adjuvant treatment: there is currently no standard for adjuvant treatment in the intermediate-risk group. Therefore, the protocol is inclusive in this aspect, allowing the choice of external beam radiotherapy with or without brachytherapy and concomitant chemotherapy. The type of adjuvant treatment is one of the stratification criteria in the protocol. (7) Adverse events and quality of life: adverse events cannot be reliably evaluated in retrospective studies. A prospective comparison of treatment-related adverse events and quality of life in both arms is therefore an important secondary endpoint in the CERVANTES trial. In addition to the standard European Organisation for Research and Treatment of Cancer (EORTC) tools for quality of life monitoring, a simple qualitative questionnaire was developed for the purpose of the study, aiming to assess the long-term impact of the treatment on health-related quality of life.
The objective of the CERVANTES trial is to provide definitive evidence (level A) for the role of adjuvant treatment in patients with intermediate-risk cervical cancer. The trial has the potential to define the new standard of care for this cohort of patients.
The first site was initiated in June 2022 and the number of trial sites is increasing progressively. The estimated date of primary endpoint analysis is 2031 and the estimated trial completion is planned for 2034.
Data availability statement
Data are available upon request.
Ethics statements
Patient consent for publication
Ethics approval
The trial was approved by the General University Hospital in Prague Ethics committee (66/20 S-MEK). Participants gave informed consent to participate in the study before taking part.
References
Footnotes
Contributors All authors planned, designed, and started the trial. DC is the trial chair and guarantor of the article. GS is the international deputy trial chair. JJ developed the statistical methods and is the trial statistician. DC and MB drafted the manuscript. All authors contributed to a critical review of the manuscript and approved the final version.
Funding This work was supported by grants from Charles University in Prague (COOPERATIO, UNCE 204065) and the Ministry of Health of the Czech Republic (MH CZ–DRO-VFN64165).
Competing interests None declared.
Provenance and peer review Commissioned; internally peer reviewed.