Article Text

Abstract
Introduction/Background*Uterine sarcoma (US) as a histologically heterogeneous group of tumours is rare and associated with a poor prognosis. This study aimed to describe the survival and identify prognostic variables in patients with US.
Methodology This unicentric, retrospective cohort study includes 57 patients with US over 18 years treated at the Department of Obstetrics and Gynaecology at University Hospital Freiburg, Germany between 1999 and 2017. Progression-free survival (PFS) and overall survival (OS) were calculated and visualised in Kaplan-Meier curves. Prognostic factors for total cohort and Leiomyosarcoma (LMS) patients were identified using log-rank test and Cox-regression.
Result(s)*44 LMS, seven low grade-endometrial stromal sarcoma (LG-ESS), four high grade-ESS and two undifferentiated US patients were identified. The median age at time of diagnosis was 51.0 years (range 18–83). The median follow-up time was 35 months. PFS for the total cohort was 14.0 months (95%-Confidence-Interval (CI) 9.7-18.3) and OS 36.0 months (95%-CI 22.1–49.9). Tumour pathology was prognostically significant for OS with LG-ESS being the most favourable (mean OS 150.3 months). In the multivariate analysis, patients over 52 years showed a four times higher risk for tumour recurrence (hazard ratio (HR) 4.4; 95%-CI 1.5-12.9). Progesterone receptor negativity was associated with a two times higher risk for death (HR 2.8; 95%-CI 1.0-7.5). For LMS patients in the univariate analysis young age (p=0.04), clear surgical margins (p=0.008), low FIGO stage (p=0.01) and no application of chemotherapy (p=0.02) were statistically significant positive factors for OS.

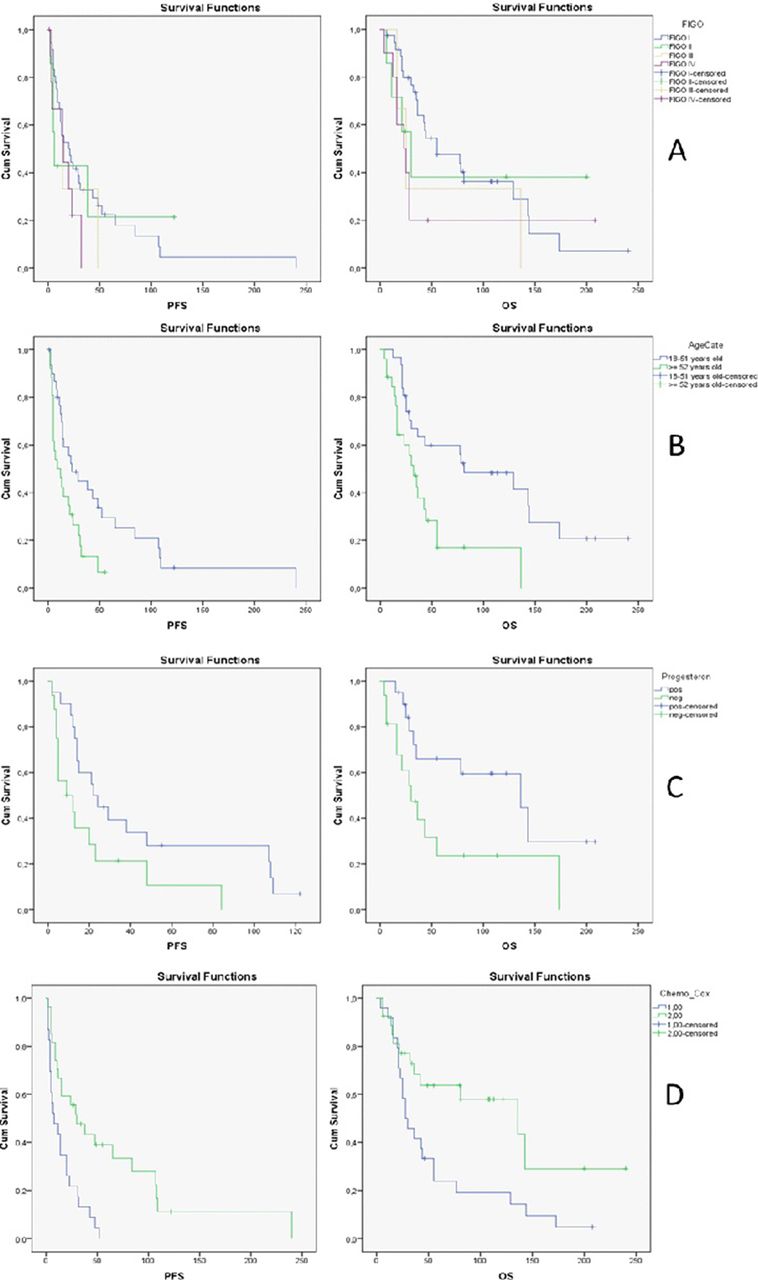{kind=link}
Kaplan-Meier curves for progression-free survival (PFS) and overall survival (OS) stratified by A: FIGO-stage, B: age category at time of diagnosis over or under 52 years, C: progesterone receptor status and D: application of chemotheraphy (1 = yes, 2 = no)
Conclusion*In this analysis, tumour histology, age at time of diagnosis and progesterone receptor status were prognostic factors for US. Unfavourable OS in LMS patients was associated with advanced FIGO stage, suboptimal cytoreduction and application of chemotherapy. In small cohorts, the confirmation in multivariate analysis stays difficult, although trends can be shown. For this aggressive but rare tumour international prospective, multicentric databases are needed to provide more concordant data. Consequent and standardised immunohistopathological workup as a basis for molecular tumour boards is worthwhile. More randomised controlled trials on adjuvant therapy are necessary to give physicians convincing treatment options especially in the recurrent situation.