Article Text

Abstract
Introduction In ARIEL3 (NCT01968213), the poly(adenosine diphosphate-ribose) polymerase inhibitor rucaparib significantly improved progression-free survival versus placebo regardless of biomarker status when used as maintenance treatment for recurrent ovarian cancer. The aim of the current analyses was to evaluate the efficacy and safety of rucaparib in subgroups based on progression-free interval following penultimate platinum, number of prior chemotherapies, and prior use of bevacizumab.
Methods Patients were randomized 2:1 to rucaparib 600 mg twice daily or placebo. Within subgroups, progression-free survival was assessed in prespecified, nested cohorts: BRCA-mutant, homologous recombination deficient (BRCA-mutant or wild-type BRCA/high genomic loss of heterozygosity), and the intent-to-treat population.
Results In the intent-to-treat population, median investigator-assessed progression-free survival was 8.2 months with rucaparib versus 4.1 months with placebo (n=151 vs n=76; HR 0.33, 95% CI 0.24 to 0.46, p<0.0001) for patients with progression-free interval 6 to ≤12 months, and 13.6 versus 5.6 months (n=224 vs n=113; HR 0.39, 95% CI 0.30 to 0.52, p<0.0001) for those with progression-free interval >12 months. Median progression-free survival was 10.4 versus 5.4 months (n=231 vs n=124; HR 0.42, 95% CI 0.32 to 0.54, p<0.0001) for patients who had received two prior chemotherapies, and 11.1 versus 5.3 months (n=144 vs n=65; HR 0.28, 95% CI 0.19 to 0.41, p<0.0001) for those who had received ≥3 prior chemotherapies. Median progression-free survival was 10.3 versus 5.4 months (n=83 vs n=43; HR 0.42, 95% CI 0.26 to 0.68, p=0.0004) for patients who had received prior bevacizumab, and 10.9 versus 5.4 months (n=292 vs n=146; HR 0.35, 95% CI 0.28 to 0.45, p<0.0001) for those who had not. Across subgroups, median progression-free survival was also significantly longer with rucaparib versus placebo in the BRCA-mutant and homologous recombination deficient cohorts. Safety was consistent across subgroups.
Conclusions Rucaparib maintenance treatment significantly improved progression-free survival versus placebo irrespective of progression-free interval following penultimate platinum, number of lines of prior chemotherapy, and previous use of bevacizumab.
- ovarian cancer
Data availability statement
Requests for de-identified datasets for the results reported in this publication will be made available to qualified researchers following submission of a methodologically sound proposal to medinfo@clovisoncology.com. Data will be made available for such requests following online publication of this article and for 1 year thereafter in compliance with applicable privacy laws, data protection, and requirements for consent and anonymization. Data will be provided by Clovis Oncology. The redacted protocol for the ARIEL3 clinical study is available on ClinicalTrials.gov (NCT01968213). Clovis Oncology does not share identified participant data or a data dictionary.
Statistics from Altmetric.com
HIGHLIGHTS
Rucaparib extended progression-free survival versus placebo regardless of penultimate progression-free interval.
Rucaparib extended progression-free survival versus placebo regardless of prior chemotherapies or bevacizumab use.
The safety profile of rucaparib was consistent across all subgroups.
Introduction
Although many patients with advanced ovarian cancer respond to initial treatment (typically surgery followed by platinum- and/or taxane-based chemotherapy), most will experience disease recurrence and require subsequent therapies.1–3 The efficacy of treatment for recurrent ovarian cancer declines rapidly with successive lines of therapy; median progression-free survival decreases from 6.4 months after the second relapse to 4.1 months after the fifth relapse,4 highlighting the need for effective therapies that delay disease progression or relapse. Historically, progression-free interval following the last dose of penultimate platinum has been used as a measure of platinum sensitivity, with an interval of ≤6 months indicating platinum-resistant disease,2 6–12 months indicating partially platinum-sensitive disease, and >12 months indicating platinum-sensitive disease.5 Although platinum sensitivity is now considered to exist on a continuum, these cut-offs are often used in clinical studies as a measure of platinum sensitivity for the purposes of selective enrolment, summarizing patient baseline characteristics, and conducting subgroup analyses.6–8
For patients with recurrent ovarian cancer who have achieved a complete or partial response to platinum-based chemotherapy, targeted agents such as poly(adenosine diphosphate-ribose) polymerase inhibitors (rucaparib, olaparib, and niraparib) and the angiogenesis inhibitor bevacizumab are routinely used as maintenance treatment, since they delay disease progression and extend the period between treatments.9–11 The choice of maintenance treatment for recurrent ovarian cancer is influenced by the treatments used in the first-line setting. Patients who did not receive a poly(adenosine diphosphate-ribose) polymerase inhibitor as first-line maintenance treatment may be eligible to receive one in the second- or later-line setting. In the USA and European Union, patients with a BRCA mutation are eligible to receive first-line olaparib maintenance treatment; those with homologous recombination deficiency may receive olaparib plus bevacizumab as first-line maintenance combination treatment,12 13 whereas first-line maintenance treatment with niraparib is approved irrespective of a patient’s BRCA or homologous recombination deficiency status.14 15 Bevacizumab may also be used as a continuation maintenance treatment in the first-line setting.16 17
In ARIEL3 (NCT01968213), the pivotal study of rucaparib maintenance treatment in recurrent ovarian cancer,18 19 rucaparib significantly improved progression-free survival versus placebo in all primary analysis groups (those with BRCA-mutant tumors, those with homologous recombination deficiency (BRCA-mutant or wild-type BRCA/high genomic loss of heterozygosity), and the intent-to-treat population). The most common any-grade treatment-emergent adverse events included gastrointestinal disorders, asthenia/fatigue, anemia/decreased hemoglobin, and dysgeusia.6 20 21 Eligible patients in this study had to be platinum sensitive (ie, documented radiological disease progression ≥6 months after the last dose of penultimate platinum).
Prognostic factors such as BRCA mutations or homologous recombination deficiency have been shown to be associated with improved efficacy from poly(adenosine diphosphate-ribose) polymerase inhibitors and from platinum-based chemotherapy. However, absence of these prognostic factors may not exclude these populations from receiving benefit; for example, rucaparib has been shown to have a progression-free survival benefit versus placebo in patients with wild-type BRCA, regardless of loss of heterozygosity status. Since progression-free interval is a known prognostic factor in ovarian cancer and a measure of platinum sensitivity,5 22 we evaluated whether rucaparib maintenance treatment was effective and safe in patients with progression-free intervals of different durations and across nested cohort subgroups based on BRCA mutation, homologous recombination deficiency, and the intent-to-treat population. In addition, as the use of 2 or ≥3 prior lines of chemotherapy, as well as the use of bevacizumab are common, the efficacy and safety of rucaparib maintenance treatment was evaluated in these settings.
Methods
Study Design
ARIEL3 is a randomized, double-blind, multicenter, phase III trial, with patients enrolled between April 7, 2014, and July 19, 2016. Comprehensive details on the study design have been published previously.6 The study was approved by national or local institutional review boards and performed in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines of the International Council for Harmonisation. Written informed consent was obtained from all patients or the requirement for written informed consent was waived by the institutional review board. In accordance with the journal’s guidelines, we will provide our data for the reproducibility of this study in other centers if requested.
Patients
Eligible patients were aged ≥18 years; had platinum-sensitive, high-grade serous or endometrioid ovarian, primary peritoneal, or fallopian tube carcinoma; had received ≥2 previous platinum-based chemotherapy regimens; had a baseline Gynecologic Cancer InterGroup cancer antigen 125 (CA-125) measurement below the upper level of normal; and achieved either a complete response according to Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST) or a partial response defined either according to RECIST or as a serological response based on CA-125 response criteria to their last platinum-based regimen. Previous treatment with bevacizumab was permitted, apart from bevacizumab maintenance after the most recent platinum-based regimen.
Procedures
Patients were stratified based on homologous recombination repair gene mutation status (based on gene mutation only; mutation in BRCA, mutation in a non-BRCA gene associated with homologous recombination, or no mutation in BRCA or a homologous recombination gene), progression-free interval following penultimate platinum-based regimen (6 to ≤12 months or >12 months), and best response to most recent platinum-based regimen (complete response or partial response), and then randomized 2:1 to receive oral rucaparib 600 mg twice daily or placebo. Patients received rucaparib or placebo in continuous 28-day cycles until disease progression (assessed using RECIST), death, or other reasons for discontinuation. Dose reductions (in decrements of 120 mg down to 240 mg) were permitted if a patient had a grade ≥3 or a persistent grade 2 adverse event. Treatment was discontinued following toxicity-related treatment interruption of >14 consecutive days. Disease assessments were conducted at screening, every 12 weeks during treatment (and after treatment for patients who discontinued for reasons other than disease progression), following clinical symptoms, and at treatment discontinuation.
Outcomes and Subgroups Analyzed
The primary outcome of ARIEL3 was investigator-assessed progression-free survival, defined as the time from randomization to investigator-assessed disease progression per RECIST or death.6 Progression-free survival was analyzed in several prespecified and post-hoc exploratory subgroup analyses using the primary efficacy data after unblinding, which was mature at a visit cut-off of April 15, 2017. Prespecified analyses of investigator- and blinded independent central review-assessed progression-free survival were conducted in subgroups defined by progression-free interval following penultimate platinum-based regimen (6 to ≤12 vs >12 months). Post-hoc analyses of investigator- and blinded independent central review-assessed progression-free survival were conducted in subgroups defined by the number of prior chemotherapy regimens (2 vs ≥3) and prior bevacizumab use (yes vs no).
Safety was assessed by monitoring for treatment-emergent adverse events classified per the Medical Dictionary for Regulatory Activities version 19.123 and graded as per the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03.24 The visit cut-off for the safety analyses was December 31, 2019.
Statistical Analyses
The rationale for target enrolment in ARIEL3 has been described previously.6 For each subgroup category, analyses were conducted in the three prespecified, nested cohorts: BRCA-mutant, homologous recombination deficient, and intent-to-treat population. Kaplan-Meier methodology was used to summarize progression-free survival; patients without documented progression were censored as of their last tumor assessment. A stratified log-rank test that included the randomization strata was used to compare treatments. Additionally, a stratified Cox proportional hazards model was used to calculate the hazard ratio (HR) between the treatment groups for progression-free survival. Treatment-by-subgroup interaction tests were performed using a Cox proportional hazards model. All efficacy endpoints were tested at a one-sided 0.025 significance level, without any multiplicity adjustment. P values for these exploratory analyses are presented for descriptive purposes only. All analyses were univariate, with no adjustment for confounding factors.
Treatment-emergent adverse events of key interest (combined alanine aminotransferase/aspartate aminotransferase elevation, combined anemia/decreased hemoglobin, combined asthenia/fatigue, nausea, combined thrombocytopenia/decreased platelet count, and vomiting) were summarized by risk difference with 95% confidence interval (95% CI). The risk difference was defined as the difference in the percentage between the rucaparib and placebo group; CIs were estimated based on normal distribution assumption. Statistical analyses were performed using SAS version 9.4 (SAS Institute, Cary, NC).
Results
Patient Characteristics
A total of 564 patients were enrolled and randomized to rucaparib (n=375) or placebo (n=189). The majority of patients had a progression-free interval of >12 months vs 6 to ≤12 months (337/564 (59.8%) vs 227/564 (40.2%)); received 2 vs ≥3 prior chemotherapy regimens (355/564 (62.9%) vs 209/564 (37.1%)); and had not received versus had received prior bevacizumab (438/564 (77.7%) vs 126/564 (22.3%); Online Supplemental Table 1). The proportion of patients who received bevacizumab in the first-line setting (71/126 (56.3%)) was similar to the proportion who received bevacizumab in the second- or later-line setting (60/126 (47.6%)). Baseline characteristics were generally well balanced between the rucaparib and placebo arms across subgroups. However, a higher proportion of patients with progression-free interval >12 months had received only two prior chemotherapy regimens (rucaparib arm: 71.4%; placebo arm: 74.3%) versus those with a progression-free interval of 6 to ≤12 months (47.0% and 52.6%). In the ≥3 prior chemotherapies subgroup, most patients had received three prior chemotherapies (rucaparib arm: 75.0%; placebo arm: 64.6%). As anticipated, patients who had received ≥3 prior chemotherapies had a shorter median time to progression on their penultimate platinum (rucaparib arm: 10.6 months; placebo arm: 11.5 months) than those who had received two prior chemotherapy regimens (16.0 and 18.0 months).
Supplemental material
Progression-Free Survival
In the intent-to-treat population, rucaparib was associated with a significant improvement in investigator-assessed progression-free survival versus placebo in patients: with a progression-free interval of 6 to ≤12 months (HR 0.33, 95% CI 0.24 to 0.46, p<0.0001) or >12 months (HR 0.39, 95% CI 0.30 to 0.52, p<0.0001); who had received two prior (HR 0.42, 95% CI 0.32 to 0.54, p<0.0001) or ≥3 prior chemotherapies (HR 0.28, 95% CI 0.19 to 0.41, p<0.0001); and who had received prior bevacizumab (HR 0.42, 95% CI 0.26 to 0.68, p=0.0004) or had not (HR 0.35, 95% CI 0.28 to 0.45, p<0.0001) (Figures 1–4). A statistically significant benefit in progression-free survival with rucaparib versus placebo was also observed across patient subgroups in the BRCA-mutant and homologous recombination deficient cohorts, and when analyzing blinded independent central review-assessed progression-free survival (Online Supplemental Figures 1–3). P values for treatment interaction tests for each subgroup were non-significant, indicating that the magnitude of treatment effect was similar regardless of progression-free interval, number of prior chemotherapy regimens, or prior bevacizumab use.

Investigator-assessed progression-free survival analyses in subgroups defined by (A) progression-free interval following penultimate platinum-based regimen, (B) number of prior chemotherapy regimens, and (C) prior bevacizumab use. P values are presented for descriptive purposes only. HRD, homologous recombination deficiency; ITT, intent to treat; PFI, progression-free interval; PFS, progression-free survival.

Investigator-assessed progression-free survival across the prespecified, nested cohorts in patients with progression-free interval following penultimate platinum-based regimen of (A–C) 6 to ≤12 months or (D–F) >12 months. P values are presented for descriptive purposes only. P values were non-significant for treatment by progression-free interval following penultimate platinum-based regimen subgroup (6 to ≤12 months vs >12 months) interaction tests (BRCA-mutant cohort, p=0.07; HRD cohort, p=0.58; ITT population, p=0.20). The dashed line indicates the median value of progression-free survival. HRD, homologous recombination deficiency; ITT, intent to treat.
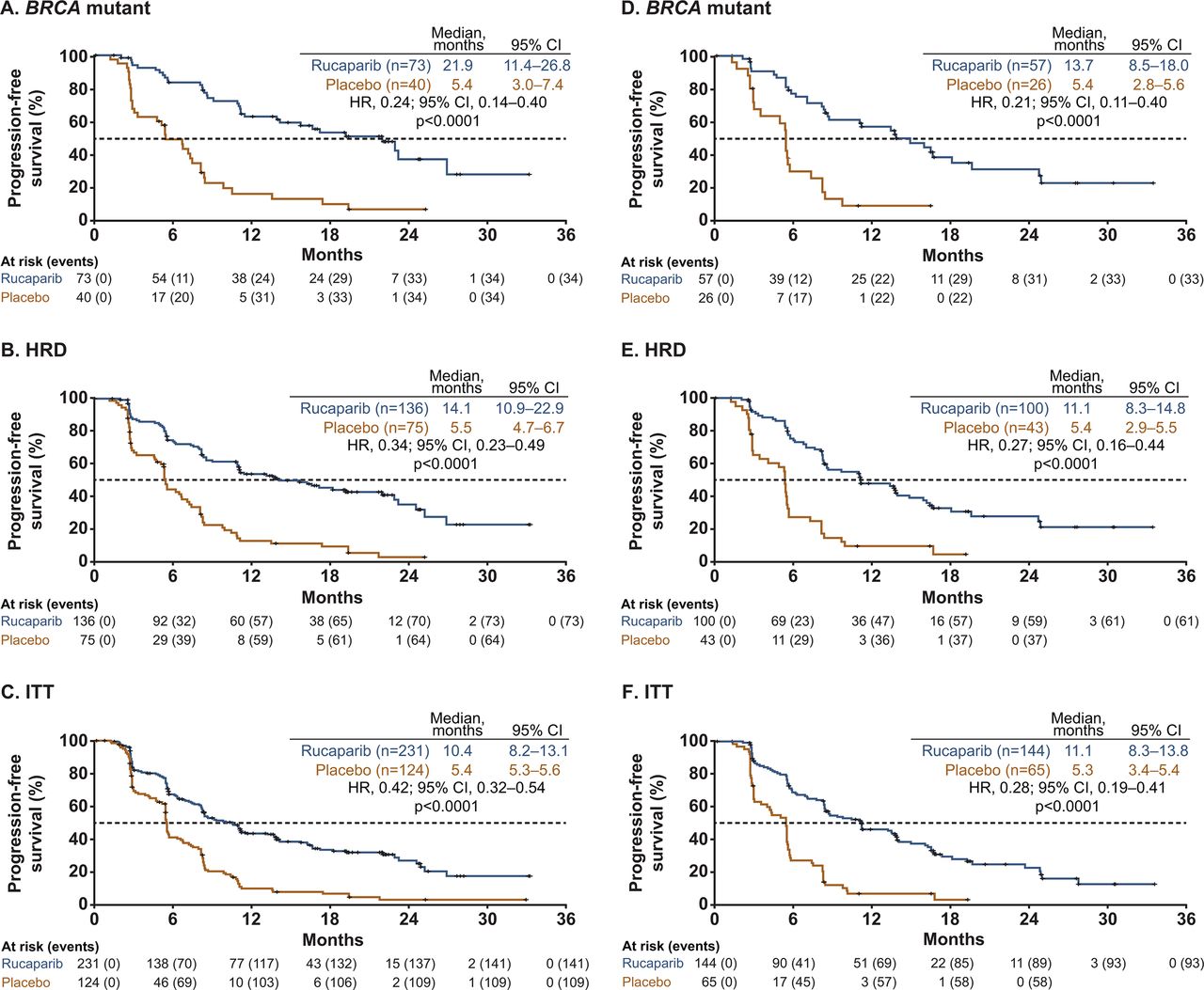
Investigator-assessed progression-free survival across the prespecified, nested cohorts in patients with (A–C) 2 prior chemotherapy regimens or (D–F) ≥3 prior chemotherapy regimens. P values are presented for descriptive purposes only. P values were non-significant for treatment by the number of prior chemotherapy regimens subgroup (2 vs ≥3) interaction tests (BRCA-mutant cohort, p=0.94; HRD cohort, p=0.65; ITT population, p=0.16). The dashed line indicates the median value of progression-free survival. HRD, homologous recombination deficiency; ITT, intent to treat.

Investigator-assessed progression-free survival across the prespecified, nested cohorts in patients with (A–C) prior bevacizumab use or (D–F) no prior bevacizumab use. P values are presented for descriptive purposes only. P values were non-significant for treatment by prior bevacizumab use subgroup (yes vs no) interaction tests (BRCA-mutant cohort, p=0.37; HRD cohort, p=0.91; ITT population, p=0.73). The dashed line indicates the median value of progression-free survival. HRD, homologous recombination deficiency; ITT, intent to treat.
In an additional exploratory analysis in patients with a progression-free interval of >24 months (Online Supplemental Figure 4), median investigator-assessed progression-free survival was significantly longer with rucaparib versus placebo (23.6 vs 6.4 months; HR 0.32, 95% CI 0.19 to 0.51, p<0.0001).
Safety
The safety population included 372/375 (99%) patients who were randomized to the rucaparib arm (three (1%) patients withdrew before receiving rucaparib) and 189 (100%) who received placebo.
In the overall safety population, the median treatment duration was 8.3 (range 0–67) and 5.5 (0–68) months in the rucaparib and placebo groups, respectively. Across subgroups, almost all patients reported ≥1 any-grade treatment-emergent adverse event; the most frequent events experienced by rucaparib-treated patients across any subgroup were nausea (75.1–78.3%) and asthenia/fatigue (68.1–78.3%) (Online Supplemental Table 2).
Any-grade hypertension did not occur at a higher rate in patients with prior bevacizumab exposure (5/83 (6.0%) and 4/43 (9.3%) in the rucaparib and placebo arms, respectively) than in those without prior bevacizumab exposure (37/289 (12.8%) and 12/146 (8.2%), respectively). Treatment-emergent adverse events associated with bevacizumab use (eg, gastrointestinal perforations and fistulae, surgery and wound-healing complications, and hemorrhage16 17) were not commonly observed in either subgroup.
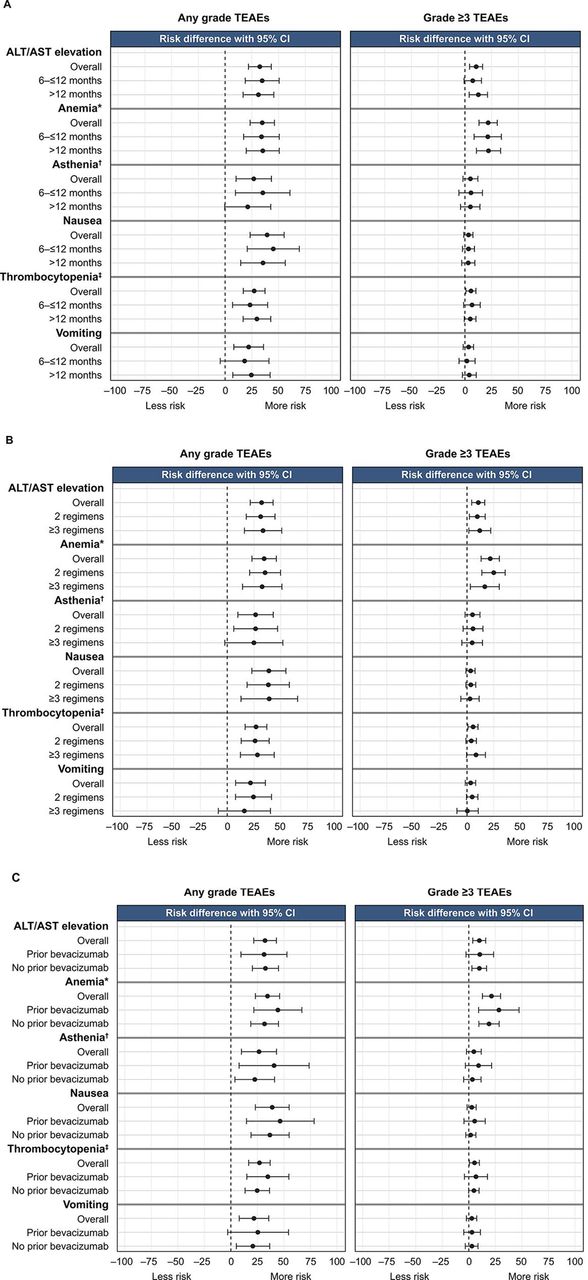
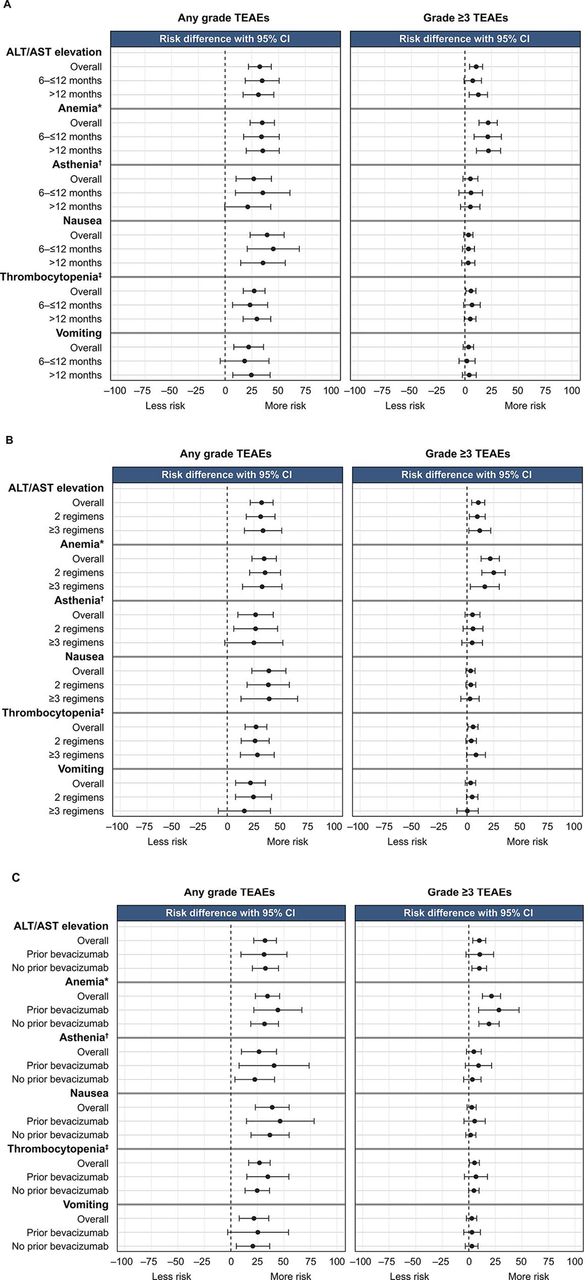
The proportion of rucaparib-treated patients who experienced grade ≥3 treatment-emergent adverse events was similar among patients with progression-free interval 6 to ≤12 months (90/150 (60.0%)) or >12 months (141/222 (63.5%)), those who had received 2 prior (142/229 (62.0%)) or ≥3 prior chemotherapy regimens (89/143 (62.2%)), and those who had received prior bevacizumab (58/83 (69.9%)) or had not (173/289 (59.9%)). Among rucaparib-treated patients, the most frequent grade ≥3 treatment-emergent adverse event across all subgroup analyses was anemia/decreased hemoglobin (Online Supplemental Table 2).
The relative risk of any-grade treatment-emergent adverse events was generally comparable across the respective subgroups and greater for patients treated with rucaparib than placebo. The relative risk of grade ≥3 anemia/decreased hemoglobin and alanine aminotransferase/aspartate aminotransferase elevations appeared higher with rucaparib than placebo in all subgroups analyzed (Figure 5).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Relative risk of any-grade and grade ≥3 treatment-emergent adverse events of key interest in subgroups defined by (A) progression-free interval following penultimate platinum-based regimen, (B) number of prior chemotherapy regimens, and (C) prior bevacizumab use. *Combined anemia and decreased hemoglobin. †Combined asthenia and fatigue. ‡Combined thrombocytopenia and decreased platelet count. ALT, alanine aminotransferase; AST, aspartate aminotransferase; TEAE, treatment-emergent adverse event.
Among rucaparib-treated patients, the proportion with treatment interruptions and/or dose reductions due to treatment-emergent adverse events was broadly similar across the three subgroups: progression-free interval (6 to ≤12 months, 106/150 (70.7%) vs >12 months, 165/222 (74.3%)); number of prior chemotherapies (2 prior, 164/229 (71.6%) vs ≥3 prior, 107/143 (74.8%)); prior bevacizumab (yes, 70/83 (84.3%) vs no, 201/289 (69.6%)). A comparable proportion of patients in the rucaparib arm of all subgroups experienced death due to treatment-emergent adverse events (excluding disease progression): none in patients with progression-free interval 6 to ≤12 months and 6/222 (2.7%) in those with progression-free interval >12 months; 3/229 (1.3%) in patients who received two prior chemotherapy regimens and 3/143 (2.1%) in those who received ≥3; 2/83 (2.4%) in patients who received prior bevacizumab and 4/289 (1.4%) in those who did not (Online Supplemental Table 2).
Discussion
Summary of Main Results
In the ARIEL3 intent-to-treat population, rucaparib maintenance treatment significantly extended median investigator-assessed progression-free survival versus placebo (10.8 vs 5.4 months, respectively; HR 0.36, 95% CI 0.30 to 0.45, p<0.0001).6 The analyses reported here add to these findings by demonstrating that rucaparib maintenance treatment significantly improved progression-free survival versus placebo in subgroups of patients with different progression-free intervals following their last platinum; in those who had received 2 or ≥3 prior chemotherapies; and in those who had and had not received prior bevacizumab. The safety profile of rucaparib was similar across all subgroups.
While patients with a progression-free interval of 6 to ≤12 months might be expected to have a worse prognosis than patients with a progression-free interval >12 months,5 our analyses indicate that rucaparib maintenance treatment provided similar benefit versus placebo across both of these subgroups. The exploratory analysis of the subgroup of patients with a prior progression-free interval >24 months, which is considered to be a highly platinum-sensitive subgroup, indicated that rucaparib also extended progression-free survival versus placebo, and that maintenance treatment may provide benefit for these patients. Although the efficacy for recurrent ovarian cancer treatments generally declines with successive lines,4 our analyses demonstrate that rucaparib provided similar benefit versus placebo in more heavily pretreated patients (ie, those who received ≥3 prior chemotherapies) and less heavily pretreated patients (ie, those who received two prior chemotherapies). Comparable efficacy was also observed in patients who previously did or did not receive bevacizumab. Overall, the safety profile in rucaparib-treated patients was similar across all subgroups and consistent with previous reports.6 25 Additional ARIEL3 subgroup analyses have also demonstrated that rucaparib is efficacious with a comparable safety profile across a range of other subgroups based on age,26 presence of bulky disease at baseline,27 and response to last platinum-based chemotherapy.28
Results in the Context of Published Literature
Our findings are consistent with analogous subgroup analyses that have been carried out for other maintenance treatments for recurrent ovarian cancer. An analysis of Study 19, a randomized phase II study of olaparib maintenance treatment in patients with recurrent ovarian cancer who received ≥2 platinum-based regimens, showed that olaparib was associated with similar efficacy versus placebo in patients with a progression-free interval of 6–12 months and >12 months.29 In the phase III SOLO2 study of olaparib maintenance treatment in patients with recurrent ovarian cancer and a BRCA mutation, there was improved progression-free survival with olaparib versus placebo regardless of number of lines of prior platinum-based chemotherapy (2, 3, or ≥4 prior lines), and prior bevacizumab did not negatively impact efficacy.8 30 In an analysis of the phase IIIb OPINION single-arm study of olaparib maintenance treatment for non-germline BRCA-mutated platinum-sensitive relapsed ovarian cancer, progression-free survival was similar among patients who received 2 or ≥3 prior platinum regimens.31 In the phase III NOVA study of niraparib maintenance in patients with platinum-sensitive recurrent ovarian cancer, niraparib maintained a benefit in progression-free survival versus placebo across subgroups when assessed by time to progression before study enrollment (6 to <12 months or ≥12 months), total number of previous platinum regimens (2 or >2), and cumulative number of previous chemotherapy regimens (2 or >2).32 Furthermore, in the phase III AURELIA study of bevacizumab as continuation maintenance in patients with platinum-resistant recurrent ovarian cancer, bevacizumab had a progression-free survival benefit versus chemotherapy alone regardless of progression-free interval (<3 months vs 3–6 months).33
Strengths and Weaknesses
One limitation of our analyses was that, although the progression-free interval subgroup analysis reported here was prespecified, the other two subgroup analyses (number of prior chemotherapies, prior bevacizumab) were post hoc in nature. In addition, our analyses were univariate rather than multivariate, with no adjustment for confounding factors. Small patient numbers in some subgroups (eg,patients receiving ≥3 prior chemotherapy regimens, or prior bevacizumab) also limit the conclusions that can be drawn for these subgroups.
Implications for Practice and Future Research
These analyses suggest that maintenance treatment is a valuable treatment option for patients with recurrent ovarian cancer from a broad range of clinically relevant subgroups. However, increasing up-front use of poly(adenosine diphosphate-ribose) polymerase inhibitors will decrease the proportion of patients with second- or later-line ovarian cancer who are naïve to these treatments. Future studies are required to determine whether patients benefit from subsequent treatment after prior exposure. Overall, these results should be regarded as hypothesis generating and could be evaluated further in appropriately designed and powered prospective studies.
Conclusions
Rucaparib significantly improved progression-free survival versus placebo in patients with a progression-free interval of 6 to ≤12 or >12 months, who had received 2 or ≥3 prior chemotherapy regimens, and who had or had not received prior bevacizumab. The magnitude of progression-free survival improvement seen with rucaparib was similar across subgroups and in the different analysis cohorts. Safety was similar between rucaparib-treated patients across all subgroups and was consistent with other reports. Taken together, these results demonstrate the consistent efficacy and safety of rucaparib maintenance treatment, even in patients who are heavily pretreated and/or have more rapid disease recurrence.
Infographic
Data availability statement
Requests for de-identified datasets for the results reported in this publication will be made available to qualified researchers following submission of a methodologically sound proposal to medinfo@clovisoncology.com. Data will be made available for such requests following online publication of this article and for 1 year thereafter in compliance with applicable privacy laws, data protection, and requirements for consent and anonymization. Data will be provided by Clovis Oncology. The redacted protocol for the ARIEL3 clinical study is available on ClinicalTrials.gov (NCT01968213). Clovis Oncology does not share identified participant data or a data dictionary.
Ethics statements
Acknowledgments
We thank all of the patients who participated in ARIEL3, as well as their families and caregivers. We also thank the investigators for their contributions to the administration and execution of the studies, and the Clovis Oncology study teams for clinical development and operational support.
References
Footnotes
ARC and DL contributed equally.
Contributors EMS, RLC, and JAL designed the study in collaboration with the funder. ARC, DL, AMO, CA, AO, AD, NC, JIW, GS, AL, RWH, MAG, PCF, JCG, DMO, DKA, SB, JG-D, EMS, RLC, and JAL treated patients. ARC, DL, AMO, CA, AO, AD, NC, JIW, GS, AL, RWH, MAG, PCF, JCG, DMO, DKA, SB, JG-D, EMS, RLC, and JAL acquired the data. All authors interpreted the data, contributed to the writing of the manuscript, reviewed and amended the drafts, and approved the final manuscript.
Funding The study was funded by Clovis Oncology, Inc. This work was supported in part by the Ann Rife Cox Chair in Gynecology and the Judy Reis/Albert Pisani, MD, Ovarian Cancer Research Fund to RLC, and National Institute for Health Research Biomedical Research Centre at University College London to JAL (no grant numbers). CA is supported in part by the Memorial Sloan Kettering Cancer Center (support grant number P30 CA008748). This work was also supported by the United States Department of Defense Ovarian Cancer Research Program OC120506, a V Foundation Translational Award, and a Stand Up To Cancer–Ovarian Cancer Research Fund Alliance–National Ovarian Cancer Coalition Dream Team Translational Research Grant (grant number SU2C-AACR-DT16–15) to EMS. Stand Up to Cancer is a program of the Entertainment Industry Foundation; research grants are administered by the American Association for Cancer Research, a scientific partner of Stand Up To Cancer. Medical writing support was provided by Andrew Croskery (Clovis Oncology, Ltd, London, UK), and editorial support funded by Clovis Oncology was provided by Frederique H Evans (Ashfield MedComms, an Ashfield Health company, Middletown, CT, USA).
Competing interests ARC has served on advisory boards for AstraZeneca, Eisai, and Tesaro/GlaxoSmithKline; has received research funding from Clovis Oncology and AstraZeneca; and has received travel and accommodation support for congress attendance from Clovis Oncology, AstraZeneca, and Roche. DL has served in a consulting or advisory role for Clovis Oncology, AstraZeneca, ImmunoGen, Merck, PharmaMar, Roche, Takeda, and Tesaro/GlaxoSmithKline, Genmab; has received institutional research support from Merck, PharmaMar and Tesaro/GlaxoSmithKline; and received support for travel or accommodation from AstraZeneca, PharmaMar, Roche, and Tesaro/GlaxoSmithKline. AMO has served on steering committees for Clovis Oncology, AstraZeneca, and Tesaro (uncompensated). CA has served on a steering committee for AbbVie and Genentech; served on advisory boards for Clovis Oncology, AbbVie, Eisai/Merck, ImmunoGen, Mersana Therapeutics, Roche, and Tesaro; and received research grants from Clovis Oncology, AbbVie, AstraZeneca, and Genentech. AO has served on advisory boards for Clovis Oncology, AstraZeneca, Genmab/Seattle Genetics, ImmunoGen, PharmaMar, Roche, and Tesaro; has received support for travel or accommodation from Clovis Oncology, AstraZeneca, PharmaMar, and Roche; and reports institutional research grant support from Clovis Oncology, AbbVie Deutschland, Ability Pharmaceuticals, Advaxis, Aeterna Zentaris, Amgen, Aprea Therapeutics, Eisai, ImmunoGen, Merck/Merck Sharp & Dohme, Millennium Pharmaceuticals, PharmaMar, Roche, and Tesaro. AD has served in a consulting or advisory role for Precision Oncology Australia, Shire Pharmaceuticals, and Specialised Therapeutics Australia. NC has served in a consulting or advisory role for Clovis Oncology, Advaxis, AstraZeneca, BIOCAD, GlaxoSmithKline, Merck Sharp & Dohme, Pfizer, PharmaMar, Roche, Takeda, and Tesaro. JIW has received research support from AbbVie and AstraZeneca and served on advisory boards for AstraZeneca. GS has served in a consulting or advisory role for Clovis Oncology, AstraZeneca, PharmaMar, Roche, and Tesaro. AL has served on advisory boards for Clovis Oncology, Ability Pharmaceuticals, AstraZeneca, BIOCAD, GamaMabs, Genmab/Seattle Genetics, Gritstone, GlaxoSmithKline, Merck Serono, Merck Sharp & Dohme, and Tesaro; steering committee for Merck Sharp & Dohme; reports institutional support for clinical trials or academic research from Clovis Oncology, Ability Pharmaceuticals, Agenus, AstraZeneca, Incyte, Inivata, Iovance, Merck Sharp & Dohme, Pfizer, Roche, Sanofi, and Tesaro; and reports boarding and travel expenses for congress activities from Clovis Oncology, AstraZeneca, and Roche. RWH has served on speakers bureaus for Clovis Oncology, AstraZeneca, and Tesaro, and on advisory boards for Clovis Oncology and AstraZeneca. MAG has served on speakers’ bureaus for Clovis Oncology, AstraZeneca, PharmaMar, and Roche. PCF has served on advisory boards for Clovis Oncology and AstraZeneca and received honoraria from AstraZeneca. JCG has received honoraria from AstraZeneca and Bristol-Myers Squibb; served in a consulting or advisory role for AstraZeneca, Bristol-Myers Squibb, GlaxoSmithKline, Merck Sharp & Dohme, and Tesaro; served on speakers’ bureaus for AstraZeneca, Ipsen, and Merck Sharp & Dohme; and received support for travel and/or accommodation from Astellas and AstraZeneca. DMO has served on advisory boards for Clovis Oncology, AbbVie, AstraZeneca, Eisai, Genentech/Roche, Genelux, Iovance Biotherapeutics, Janssen, Novocure, Regeneron, and Tesaro; has served on steering committees for Clovis Oncology, Agenus, Amgen, and Novocure; has served as a consultant for AbbVie, Ambry, AstraZeneca, Genentech/Roche, Gynecologic Oncology Group Foundation, and Tesaro; has given a presentation on ovarian cancer at the National Comprehensive Cancer Network; and his institution has received research support from Clovis Oncology, AbbVie, Agenus, Ajinomoto, Amgen, Array BioPharma, AstraZeneca, Bristol-Myers Squibb, Cerulean Pharma, Eisai, EMD Serono, ERGOMED Clinical Research, Genentech, Gynecologic Oncology Group, INC Research, inVentiv Health Clinical, Iovance Biotherapeutics, Janssen Research and Development, Ludwig Institute for Cancer Research, New Mexico Cancer Care Alliance, Novocure, PRA International, Regeneron Pharmaceuticals, Serono, Stemcentrx, Tesaro, TRACON Pharmaceuticals, VentiRx, and Yale University. DKA has served as a scientific advisor for Morphotek and received research funding from Clovis Oncology, Advaxis, AstraZeneca, Pfizer, Syndax, and Tesaro. SB has served on advisory boards and received honoraria from Clovis Oncology, AstraZeneca, Genmab, GlaxoSmithKline, Immunogen, Merck Sereno, Merck Sharp & Dohme, Mersana, Pfizer, Roche, Seattle Genetics, and Tesaro; received honoraria for lectures from AstraZeneca/Merck Sharp & Dohme, GlaxoSmithKline, Pfizer, Roche, and Tesaro; received support for travel or accommodation from NuCana and Tesaro; and reports institutional funding from AstraZeneca, GlaxoSmithKline, and Tesaro. JG-D has received research funding from AstraZeneca, Pierre Fabre, and Pfizer; received personal fees from Clovis Oncology, Astellas, Pierre Fabre, and Pfizer; and received nonfinancial support from Astellas, Pierre Fabre, and Pfizer. TC and SG are employees of Clovis Oncology and may own stock or have stock options in that company. RLC reports grants from Clovis Oncology, AstraZeneca, Gateway Foundation, Janssen, Judy Reis/Albert Pisani, MD, Ovarian Cancer Research Fund, Merck, National Institutes of Health, Roche/Genentech, and V-Foundation; has served as an advisor to Clovis Oncology, Agenus, AstraZeneca, GamaMabs, Genmab, Janssen, OncoQuest, Pfizer (Medivation), Regeneron, Roche/Genentech, and Tesaro; and has an endowment as the Ann Rife Cox Chair in Gynecology. JAL has received lecture fees from Clovis Oncology, AstraZeneca, and Pfizer; served on advisory boards for Clovis Oncology, Artios Pharma, AstraZeneca, Cristal Therapeutics, Merck/Merck Sharp & Dohme, Pfizer, Regeneron, Roche, Seattle Genetics, and Tesaro; and received research grants from AstraZeneca and Merck/Merck Sharp & Dohme.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.