Article Text

Abstract
Elevated levels of replicative stress in gynecological cancers arising from uncontrolled oncogenic activation, loss of key tumor suppressors, and frequent defects in the DNA repair machinery are an intrinsic vulnerability for therapeutic exploitation. The presence of replication stress activates the DNA damage response and downstream checkpoint proteins including ataxia telangiectasia and Rad3 related kinase (ATR), checkpoint kinase 1 (CHK1), and WEE1-like protein kinase (WEE1), which trigger cell cycle arrest while protecting and restoring stalled replication forks. Strategies that increase replicative stress while lowering cell cycle checkpoint thresholds may allow unrepaired DNA damage to be inappropriately carried forward in replicating cells, leading to mitotic catastrophe and cell death. Moreover, the identification of fork protection as a key mechanism of resistance to chemo- and poly (ADP-ribose) polymerase inhibitor therapy in ovarian cancer further increases the priority that should be accorded to the development of strategies targeting replicative stress. Small molecule inhibitors designed to target the DNA damage sensors, such as inhibitors of ataxia telangiectasia-mutated (ATM), ATR, CHK1 and WEE1, impair smooth cell cycle modulation and disrupt efficient DNA repair, or a combination of the above, have demonstrated interesting monotherapy and combinatorial activity, including the potential to reverse drug resistance and have entered developmental pipelines. Yet unresolved challenges lie in balancing the toxicity profile of these drugs in order to achieve a suitable therapeutic index while maintaining clinical efficacy, and selective biomarkers are urgently required. Here we describe the premise for targeting of replicative stress in gynecological cancers and discuss the clinical advancement of this strategy.
- ovarian cancer
- medical oncology
- uterine cancer
- cervical cancer
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, an indication of whether changes were made, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
The relevance of replicative stress in cancer
Replicative stress and its cellular consequences
Maintaining genomic integrity is of utmost importance to eukaryotic cells, which have evolved sophisticated mechanisms to ensure speed, accuracy, and an adequate pool of nucleotide and replication factors as well as high-fidelity repair pathways to correct errors occurring during DNA replication. Replication of DNA is initiated at multiple sites along the genome, known as replication origins, which form bidirectional replication forks. Each origin is initiated by a combination of regulatory proteins that prepare the chromatin for replication before synthesis (S)-phase entry. In the presence of errors or damage during DNA replication, cell cycle checkpoint nodes and repair machinery work in concert to retard cell cycle progression until sufficient repair has been achieved. Any obstacles encountered by cells in this process can lead to ‘replicative stress’ (Figure 1),1 which may be overcome by replicative stress response proteins, but deficiencies in this response result in accumulated errors in DNA replication and loss of genomic integrity, which lead to cell death.
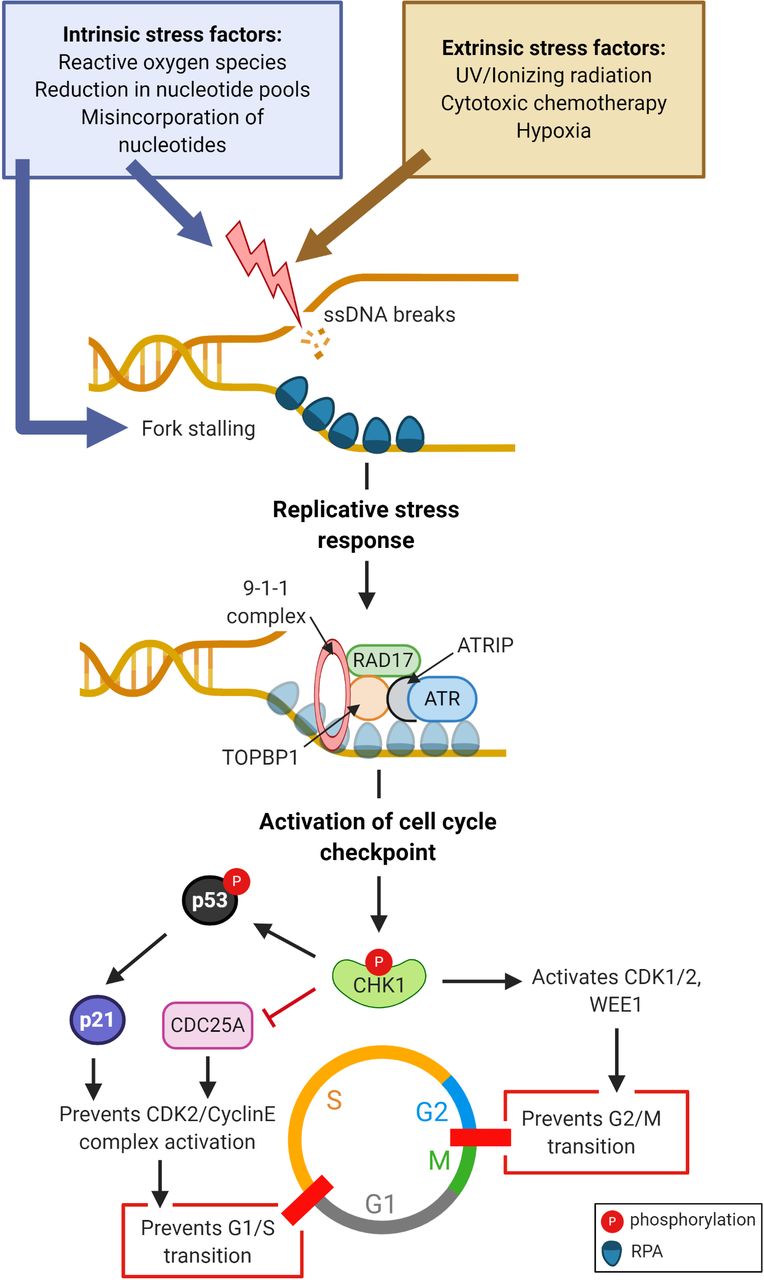
Onset of replicative stress and subsequent signaling events. Exogenous and endogenous factors trigger formation of single-stranded DNA (ssDNA) breaks and/or stalling of replication forks, and recruit replication protein A (RPA), which commence the replicative stress response. RPA-coated ssDNA recruits the assembly of ataxia telangiectasia and RAD17 protein complexes. Ataxia telangiectasia and Rad3 related (ATR) kinase phosphorylates checkpoint kinase 1 (CHK1), which prevents G1/S and G2/M transition through suppression of cell division cycle 25 (CDC25A) and activation of cyclin dependent kinase (CDK1/2), respectively, leading to cell cycle arrest. CHK1 and CHK2 phosphorylate and stabilize p53, p21 and Rb, which maintain cell cycle arrest. Furthermore, CHK1 phosphorylates and activates the negative regulator of CDK1/2, WEE1-like protein kinase (WEE1). (Figure created with Biorender.com).
The replicative stress response ultimately activates the DNA damage sensor ataxia telangiectasia and Rad3 related (ATR) kinase which triggers the DNA damage response cascade, culminating in the activation of key cell cycle checkpoints, initiation of DNA repair, and restoration of stalled forks2 (Figure 1). The ATR checkpoint kinase 1 (CHK1) signaling pathway is the major organizer of the replicative stress response by inducing cell cycle arrest, suppressing global origin firing, and interrupting phase-transition checkpoints, essentially providing a ‘breather’ for cells to recover. Cell cycle arrest is effected by CHK1-mediated suppression of M-phase inducer phosphatase 1/3 (CDC25A/C), marking them for ubiquitination and degradation (Figure 1). By modulating the activity of CHK1, ATR is therefore a major regulator of the Gap 1 (G1)/S and Gap 2 (G2)/Mitotic (M) checkpoints, preventing cells with damaged DNA from progressing further towards mitosis.2
ATR activation also mediates protection, reversal, and restart of stalled forks. Fork reversal is facilitated by the cooperation of RAD51 recombinase (RAD51) with fork remodelers such as Zinc Finger RANBP2-Type Containing 3 (ZRANB3) and SWI/SNF-related, matrix-associated, actin dependent regulator of chromatin, subfamily-a-like-1 (SMARCAL1). This prevents single-stranded breaks from collapsing into double-stranded DNA breaks. BRCA2 and poly (ADP-ribose) polymerase 1 (PARP1) independently protect stalled forks by loading DNA repair protein RAD51 homolog 1 (RAD51) at the fork, preventing cellular nucleases such as crossover junction endonuclease MUS81 (MUS81) and double-strand break repair protein MRE11 (MRE11) from digesting exposed DNA, which could lead to fork collapse.3 WEE1-like protein kinase (WEE1) further protects forks by negatively regulating MUS81.3 Subsequently, fork restart occurs through homologous recombination, trans-lesion synthesis, and break-induced replication.4 Otherwise, collapse of the fork into double-stranded DNA breaks, in the presence of underlying deficiencies of double-stranded DNA breaks repair, may drive the cell towards apoptosis or senescence.4
In cancer cells, the abrogation of cell cycle checkpoints resulting from mutations in P53, cyclins or other tumor suppressors during carcinogenesis and cancer progression, as well as the oncogenic signals that accelerate cell cycling and proliferation, may be the Achilles’ heel for therapeutic exploitation. Given the pleiotropic roles of ATR, inhibition of the ATR–CHK1–WEE1 axis provides the opportunity to enhance replicative stress through fork destabilization while further lowering cell cycle checkpoint thresholds, thus hurtling tumor cells toward the point of mitotic catastrophe and cell death.
Exogenous and endogenous sources of replicative stress
Cytotoxic chemotherapies are a major exogenous source of replicative stress for cancer cells. Cisplatin and carboplatin are alkylating agents forming intra- and inter-strand crosslinks between bases that affect accurate base pairing and disrupt unwinding of DNA strands, respectively.5 Gemcitabine, a nucleoside analog, reduces intra-cellular nucleotide pools required for DNA synthesis, triggering replicative stress by reducing the speed of fork progression. Aberrant integration of nucleoside analogs during DNA synthesis may further terminate DNA chains. Similarly, methotrexate, another anti-metabolite, inhibits dihydrofolate reductase, thus reducing the rate of DNA synthesis. Topoisomerase II inhibitors such as pegylated liposomal doxorubicin bind to DNA strands and cause transient double-stranded breaks which are unable to undergo re-ligation, activating CHK1,6 while topoisomerase I inhibitors induce replication blocks by generating topoisomerase I–DNA cleavage complexes. Anti-angiogenics such as bevacizumab, particularly in the context of haphazard tumorous vasculature, can exacerbate hypoxic and nutrient-deprived tumor microenvironments, reducing raw materials for nucleotide production, increasing replicative stress. Hypoxia is also associated with reduced ribonucleotide reductase activity and increased mitochondrial reactive oxygen species, which further contribute to replicative stress.
Endogenously, uncontrolled oncogenic activation, such as via the upregulation of RAS, C-MYC, and CCNE1, is an important source of replicative stress, resulting in the accumulation of genomic instability. Oncoproteins may stimulate premature G1/S progression, triggering S-phase in cells lacking sufficient DNA replication machinery, while amplification of CCNE1 increases cyclin E levels leading to aberrant firing of the replication origin. Increased MYC activity has links with defective reduction/oxidation balance in cells, and an accumulation of reactive oxygen species which induce replicative stress by the formation of oxidized nucleotides such as 8-oxoguanine, leading to mismatched base pairing.7 Similarly, mutations in gatekeeper tumor suppressor genes that regulate cell cycle checkpoints, such as in P53 and RB1, allow early G1/S transition, increasing genomic instability. Compounding this, deficiencies in homologous recombination and mismatch DNA repair pathways lead to increased common fragile sites4 and reduced fork repair, contributing toward replicative stress.
Rationale for targeting replicative stress in gynecological cancers
High grade serous carcinoma represents 70–80% of diagnosed ovarian cancer and is characterized by marked genomic instability.8 Half of high grade serous carcinoma tumors harbor defects in homologous recombination DNA repair, and amplification of CCNE1 (20%), RB1 loss (15%), as well as mutations in ATR (2%) and ATM (2%) are not infrequent.9 Furthermore, P53 is ubiquitously mutated in high grade serous carcinoma, increasing their reliance on the G2/M checkpoint. Targeting cell cycle checkpoints through inhibition of the ATR–CHK1–WEE1 axis may therefore induce synthetic lethality in high grade serous carcinoma cells with oncogenic stress or which harbor intrinsic deficiencies in DNA repair.
The increasing number of approvals for PARP inhibitors (PARPis) in advanced ovarian cancer therapy indicates that PARPis are steadily shifting treatment paradigms, heralding an increasing proportion of patients who are at risk of PARPi-resistant disease. PARPi resistance occurs through several independent mechanisms that have been grouped into three categories: (1) mitigation of replication stress by replication fork protection, such as through the loss of mixed-lineage leukemia protein 3/4 (MLL3/4) complex protein Pax2 transactivation domain interacting protein (PTIP) which prevents MRE11 from being recruited to stalled forks;10 (2) restoration of homologous recombination activity; and (3) processes that do not fall under any single DNA repair pathway but alter the response to PARPi, such as increased drug efflux, loss of PARP1 expression, and down-regulation of PARP trapping capacity.11 In PARPi-resistant but BRCA-mutant ovarian cancer cells, both homologous recombination and fork protection functions of BRCA1/2 are sequentially bypassed and cells become increasingly dependent on ATR for recruitment of RAD51 onto double-stranded breaks and stalled forks.12 13 Inhibition of ATR using the ATR inhibitor (ATRi) VE-821 in olaparib-resistant BRCA1-mutant UWB1.289 ovarian cancer cells reduced RAD51 loading and prevented BRCA1-independent fork protection, leading to increased MRE11-mediated degradation of stalled forks. These effects resulted in re-sensitization of olaparib-resistant UWB1.289 ovarian cancer cells to olaparib.12 In another study of BRCA1-mutant ovarian cancer cell lines and patient-derived xenograft models with acquired olaparib resistance, the CHK1 inhibitor (CHK1i) prexasertib was able to restore homologous recombination and stabilize replication forks.14 Together, this suggests that inhibiting the ATR–CHK1–WEE1 axis holds promise to reverse PARPi resistance in ovarian cancer.
Further genotypic contexts relevant to gynecological cancers, where ATR–CHK1–WEE1 inhibition has potential synthetic lethality, include cells with overexpression of RAS, CCNE1 amplification, and ARID1A mutation. CCNE1 overexpression prompts early S-phase entry and increases genomic instability, increasing reliance on homologous recombination DNA repair. RAS-transformed cells demonstrate increased reliance on the ATR–CHK1 axis, and ATR inhibition in this context led to increased genomic instability and resultant synthetic lethality.15 ARID1A mutations occur in ~50% of ovarian and endometrial clear cell carcinoma and ~30% of endometrial cancers of endometrioid and carcinosarcoma subtype. After DNA damage, AT-rich interacting domain containing protein 1A (ARID1A) assists in non-homologous end-joining (NHEJ) DNA repair by recruiting x-ray repair cross-complementing 5 and 6 (XRCC5/6) to sites of double-stranded breaks, acts as a binding partner of ATR, and sustains DNA damage signaling in response to double-stranded breaks.16 Using genetic screens, Williamson identified ARID1A as a synthetic lethal partner for ATR inhibition and showed in-vitro susceptibility to ATRi in a variety of histologically diverse ARID1A-mutant cell lines. In TOV21G ARID1A-mutant ovarian clear cell carcinoma xenograft models, sensitivity to the ATRi berzosertib was shown and was associated with features suggesting mitotic catastrophe, such as the accumulation of cells in G2/M, and elevated markers of chromosomal instability and apoptosis induction.17
Finally, in cervical cancer, high-risk human papilloma virus (HPV) infection is implicated in >70% of cases. Cells infected with high-risk HPV strains show constitutive activation of ataxia telangiectasia-mutated (ATM), and to a lesser degree ATR, throughout the viral life cycle, even in the absence of exposure to DNA-damaging radiation or chemotherapy.18 Supporting this, levels of downstream members phosphorylated-ATR, CHK1/2, and BRCA1 have been shown to be elevated in high-risk HPV-infected cells.18 Furthermore, in HPV-infected cells, the p53 checkpoint is often inactivated by E6-mediated ubiquitination and proteasomal degradation, which could lead to dependence on other cell cycle checkpoints. Although the exact mechanisms through which activation of the ATM pathway leads to viral amplification in differentiated cells still remain unclear, these links are provocative for therapeutic targeting of replicative stress in cervical cancer.
Targeting replicative stress in ovarian cancer: clinical advances
Targeting the DNA damage sensors
ATR inhibition
The broad role of ATR in DNA damage sensing, fork protection, and the DNA damage restriction checkpoint lends to its immense potential for inhibition in cancer therapy. Four potent and highly selective ATRis—M6620, AZD6738, BAY1895344, and M4344—are currently in clinical development (Table 1) (Figure 2).

{kind=link}
{kind=link}
DNA replication stress and clinical grade inhibitors of relevant cell cycle checkpoints. SSBs, single-strand breaks; DSBs, double-strand breaks; ssDNA, single-strand DNA; BER, base-excision repair; PARP, poly(ADP-ribose) polymerase; XRCC1/4, X-ray repair cross-complementing protein 1 and 4; ATR, ataxia telangiectasia and Rad3-related; CHK1/2, checkpoint kinases 1 and 2; CDC25 A and C, M-phase inducer phosphatase 1 and 3; CDK, cyclin-dependent kinase; WEE1, WEE1-like protein kinase; ATM, ataxia telangiectasia mutated; RNR, ribonucleotide reductase; NHEJ, non-homologous end-joining; DNA-PK, DNA-dependent protein kinase; HR, homologous recombination (Figure created with Biorender.com).
Classes of therapy targeting replication stress in gynecological cancers
Berzosertib (M6620, VX-970, Merck) is a first-in-class ATRi. In a phase I study of advanced solid tumors treated with berzosertib ± carboplatin, berzosertib monotherapy was well tolerated with no high-grade toxicities or dose-limiting toxicities (DLT).19 However, when combined with chemotherapy, frequent hematological DLT necessitated dose interruptions and reductions. Berzosertib treatment was associated with a reduction in phosphorylated-CHK1 levels19 on paired tumor biopsies pre- and post-treatment, confirming its expected pharmacodynamic effect. Although limited monotherapy activity was reported (objective response rate of 9%), complete response exceeding 19 months was noted in a colorectal cancer patient with ATM loss, and a durable partial response was seen in a patient with BRCA1-mutant but platinum- and PARPi-resistant high grade serous carcinoma harboring somatic mutant-P53. In another phase I study of berzosertib plus veliparib and cisplatin, a durable partial response occurred in a BRCA-wildtype platinum-resistant patient with ovarian cancer.20 Currently, combinatorial approaches with chemotherapy are under investigation in high grade serous carcinoma (Table 1). Recently, a randomized phase II study of gemcitabine ± berzosertib in platinum-resistant high grade serous carcinoma has reported improvements in progression-free survival for the combination arm. Interestingly, progression-free survival benefit occurred only in the subgroup of patients who had a short platinum-free interval of <3 months; no progression-free survival benefit was seen in patients with a longer platinum-free interval21 (Table 1).
Ceralasertib (AZD6738, AstraZeneca) is a selective and potent oral ATRi. As monotherapy, ceralasertib showed an objective response rate of 7% in a phase I study of advanced solid tumors; however, 48% of patients achieved stable disease, with durable responses noted (NCT02223923). Continuous daily dosing was not tolerated due to cumulative myelosuppression, thus different dosing schedules are being explored in future expansion cohorts which will focus on selecting patients with known homologous recombination deficiency. Study 4 was a phase I trial combining ceralasertib with carboplatin or olaparib in advanced solid tumors. Unsurprisingly, the combination of ceralasertib with carboplatin was not well tolerated due to myelosuppression. In this cohort, 3/37 responses were seen, including one patient with ATM-mutant clear cell ovarian cancer (NCT02264678).
ATM inhibition
ATM has a key role in activating the DNA damage response in response to double-stranded breaks (Figure 2). In pre-clinical models, the selective ATM inhibitor AZD0156 (AstraZeneca) was a good radiosensitizer in-vitro and in-vivo.22 The combination of AZD0156 with olaparib impaired the ability of cells to repair olaparib-induced DNA damage, which culminated in increased double-stranded breaks and cell cycle arrest.22 A phase I study of AZD0156 in combination with olaparib or chemotherapy in advanced solid tumors is ongoing (NCT02588105).
Targeting efficient cell cycle modulation
Disrupting the G2/M cell cycle checkpoint: WEE1 inhibitors
The inhibition of WEE1 increases cyclin-dependent kinase (CDK) 1 and 2 activity, abrogating the G2/M checkpoint and allowing cells to inappropriately enter mitosis with damaged DNA (Figure 2). This subsequently triggers a spike in cell cycling and depletes nucleotide pools, further increasing replicative stress. As a result, more replication forks are stalled, and new double-stranded breaks are produced from endonuclease activity.11 WEE1 has also been implicated in homologous recombination DNA repair, and its inhibition hampers homologous recombination repair through CDK1-mediated BRCA1/2 phosphorylation.23 In ovarian cancer, WEE1 was found to be overexpressed in 92% of effusions from advanced high grade serous carcinoma,24 and expression was significantly higher in chemotherapy-refractory compared with treatment-naive patients. High WEE1 expression correlated independently with a worse overall survival.24 These data have bolstered the rationale for WEE1 inhibition in tumors that are p53-deficient and are therefore more reliant on the G2/M checkpoint. Pre-clinical studies of the WEE1 inhibitor (WEE1i) adavosertib (AZD1775, MK 1775; AstraZeneca) corroborate that WEE1i sensitizes P53-null tumors to radiation and cytotoxic chemotherapy by increasing mitotic catastrophe.25
Currently, adavosertib is the only WEE1i in clinical development. In a phase I study of adavosertib monotherapy, 2/10 patients achieved clinical benefit, both of whom were high grade serous carcinoma patients with germline BRCA1/2 mutations.26 Retrospective analysis of all tumor samples showed that 5/10 patients had tumors harboring P53 mutations, but none of the patients responded despite the suggested rationale for synthetic lethality with WEE1 inhibition in this context.26 Another randomized phase II study has reported the combination of adavosertib, carboplatin and paclitaxel versus chemotherapy alone in platinum-sensitive relapsed P53-mutant ovarian cancer, showing improvements in outcomes for the adavosertib combination27 (Table 1). In recurrent platinum-resistant ovarian cancer, a randomized phase II study has evaluated the combination of gemcitabine ± adavosertib in patients with heavily pre-treated platinum-resistant ovarian cancer based on pre-clinical models suggesting a synergistic increase in replicative stress after gemcitabine treatment. Preliminary data have described shown improved progression-free survival and overall survival with adavosertib combination therapy28 (Table 1). However, the incidence of high grade myelosuppression was doubled in patients receiving adavosertib plus gemcitabine compared with gemcitabine alone. In another phase II study, adavosertib was combined with carboplatin, gemcitabine, paclitaxel or pegylated doxorubicin29 (Table 1). The highest objective response rate was noted with the combination of adavosertib and carboplatin, and median progression-free survival in this cohort was 10 months but was associated with high rates of grade ≥3 myelosuppression; 63% of patients required a dose reduction of adavosertib29 (Table 1). This study suggested a possible positive correlation between CCNE1 amplification and adavosertib response, and a phase II study specifically recruiting patients with CCNE1 amplification is currently open (Table 1). The outcome of this trial would be of great interest given that CCNE1 amplification occurs in around 20% of high grade serous carcinoma and is associated with primary and acquired chemotherapy resistance and a poor prognosis.9
CHK 1 and 2 inhibitors
CHK1i increase replicative stress by suppressing the repair of stalled forks and by depleting nucleotide pools through aberrant replication origin firing, while allowing the G2/M transition to proceed (Figure 2). Prexasertib (LY2606368; Eli Lilly) is an intravenous CHK1i with minor activity against checkpoint kinase 2 (CHK2). In a phase II proof-of-concept study of 28 women with BRCA1/2-wildtype platinum-resistant high grade serous carcinoma or endometrioid ovarian cancer receiving prexasertib, the partial response rate was 29%.30 Major side effects were myelosuppression, with 79% of patients experiencing transient grade 4 neutropenia30 (Table 1). Further analysis revealed that 12/19 women (63%) whose tumors harbored pre-therapy CCNE1 amplification, overexpression, or both, experienced durable progression-free survival >6 months30 (Table 1). In a first-in-human trial of an oral CHK1i, SRA737 (Sierra Oncology) monotherapy in advanced solid tumors, patients with heavily pre-treated high grade serous carcinoma experienced a favorable disease control rate of 54%; however, no convincing association with sensitivity could be demonstrated in the small subset of patients with CCNE1 amplification. Subjects with mutations in the Fanconi anemia/BRCA network had the most favorable outcomes (disease control rate 71% in this group)31 (Table 1).
Targeting cell cycle coordination: CDK inhibitors
Cyclins and their paired CDKs control cell cycle progression through their sequential activation and quiescence (Figure 2). CDK targeting therefore has the potential to contribute towards increasing replicative stress, and is increasingly being evaluated in cancer therapy. In patients with recurrent epithelial ovarian cancer demonstrating RB-proficiency and low p16 expression, palbociclib, a CDK4/6 inhibitor, showed limited monotherapy activity32 (Table 1). Currently, the utility of CDK 4/6 inhibition in high grade serous carcinoma remains unclear, especially given known ubiquitous p53 dysfunction, which may limit the benefit of this approach.33 Conversely, low grade serous ovarian cancer is known to have moderate sensitivity to endocrine therapy, and hormonal manipulation similar to that used in estrogen-receptor positive advanced breast cancer has been used. Combinations of CDK4/6 inhibitors with endocrine therapy in low grade serous ovarian cancer are under investigation (Table 1).
Dinaciclib, an inhibitor of CDK 1, 2, 5 and 9, has shown potential to reverse PARPi resistance in BRCA1-mutant but PARPi-resistant breast cancer cell lines and patient-derived xenografts.34 Dinaciclib reduced RAD51 expression and, when combined with PARPi, resulted in increased ϒ- H2A histone family member X (H2AX) foci compared with PARPi treatment alone.34 Phase I studies are ongoing (Table 1). Finally, a highly potent and selective CDK7 inhibitor, SY-1365 (Syros), strongly and specifically downregulated cell cycle as well as homologous recombination repair and mismatch DNA repair-related genes when tested in ovarian cancer xenografts.35 A phase I study is investigating SY-1365 in advanced breast and ovarian tumors (Table 1). We await the results of these studies to better define the role for CDK inhibition in ovarian cancer treatment.
Impeding DNA repair to accentuate replicative stress
Targeting non-homologous end-joining repair of double-strand breaks
DNA-dependent protein kinase (DNA-PK) has key roles in initiating NHEJ (Figure 2) and in regulating cell cycle progression. The catalytic subunit of DNA-PK (DNA-PKcs) phosphorylates p53 and reduces mouse double minute two homolog (MDM2) binding, thus stabilizing p53 and reducing G1/S transition. DNA-PKcs also phosphorylates replication protein A (RPA), leading to cell cycle arrest and delaying mitosis.2 Homologous recombination DNA repair deficiency may be a predictive marker for synthetic lethality with DNA-PKcs inhibition, given the increased reliance of homologous recombination-deficient cells on NHEJ to maintain genomic integrity.11 Pre-clinical studies have demonstrated that AZD7648 (AstraZeneca), a DNA-PKcs inhibitor, increased genomic instability in combination with olaparib in ATM-deficient cells, increasing apoptosis and leading to sustained tumor regression in-vivo.36 Nedisertib (M3814;Merck) is another selective orally bioavailable DNA-PKcs inhibitor. Only limited monotherapy activity was noted in P53-wildtype ovarian cancer xenografts, however nedisertib plus pegylated liposomal doxorubicin showed improved activity compared with pegylated liposomal doxorubicin alone in-vivo. Currently, a phase Ib study of nedisertib in combination with pegylated liposomal doxorubicin is planned, with expansion into cohorts of ovarian cancer (Table 1).
Inhibiting ibonucleotide reductase to suppress deoxyribonucleotide supply
Ribonucleotide reductase is required for de-novo deoxyribonucleotide synthesis which is required in abundance for repair of stalled forks. The potent small-molecule ribonucleotide reductase inhibitor triapine (3-AP) effectively prevents deoxyribonucleotide synthesis, enhancing replicative stress (Figure 2). In pre-clinical studies, triapine disrupted homologous recombination repair, thus sensitizing BRCA-wildtype ovarian cancer cells to further DNA damage from doxorubicin, cisplatin, and PARPi, synergistically.37 In PARPi-resistant ovarian cancer cells, the combination of olaparib, triapine, and cediranib was able to reverse PARPi resistance in-vitro.38 An objective response rate of 17% and median overall survival of 7 months was observed in six patients with platinum-resistant ovarian cancer who were treated with overlapping triapine with cisplatin in a phase I study. However, one-third of the patients developed triapine-related methemoglobinemia due to the disruption of collateral proteins containing iron by the ribonucleotide reductase inhibitor, and the trial was discontinued (Table 1).
Targeting replicative stress in combination with PARPi
PARPis inhibit single-stranded DNA break repair by limiting the base excision repair pathway, but also form toxic PARP-DNA complexes via PARP trapping which block DNA replication, increase replicative stress, induce ATR activation, and trigger the S-phase checkpoint. Rational combinations of PARPi with ATRi–CHK1i–WEE1i are being explored based on the premise of synthetic lethality, as the replicative stress induced by PARPi requires downstream DNA damage response signaling for resolution. This may expand the role of PARPi into treatment paradigms for homologous recombination-proficient high grade serous carcinoma.
In both BRCA1-mutant PEO1 and BRCA2-reversion PEO4 ovarian cancer cells, the combination of olaparib with the ATRi ceralasertib (AZD6738) was shown to be synergistic compared with PARPi monotherapy and led to increased markers of chromosomal instability and apoptosis.13 This synergism was attributed to dual inhibition of independent fork-stabilizing mechanisms controlled by ATR/CHK1 and PARP, as well as increased mitotic catastrophe due to premature mitotic entry of cells harboring high levels of double-stranded breaks.13 Comparatively, varying degrees of genomic instability and tumor survival were induced by olaparib/ceralasertib and olaparib/MK8776 (a CHK1i) combinations in-vitro and in-vivo. The olaparib/ceralasertib combination appeared to provide superior tumor regression in BRCA-mutant patient-derived xenograft models, while the olaparib/MK8776 combination was cytostatic but failed to induce tumor shrinkage.13 Therefore, the distinct functions of ATR and CHK1 may affect their therapeutic efficacy in combination with PARPi. The combination of concurrent PARPi plus WEE1i has been shown to be similarly synergistic in ovarian cancer models across different histological and genomic profiles,39 leading to increased DNA damage, mitotic catastrophe, and apoptotic death. For example, in talazoparib-resistant OVCAR8 xenograft models only a modest adavosertib response was noted, but marked growth inhibition occurred with treatment with the combination of adavosertib plus PARPi.39 Pre-clinical data also suggest that the degree of synergy between PARPi and DNA damage response pathway inhibitors may depend on the PARP1-trapping potency of the specific PARPi, as higher PARP1-trapping affinity generates higher levels of replicative stress from increased PARP–DNA complexes which are highly disruptive to DNA replication. The choice of sequential or concurrent pathway inhibition may also depend on the degree of endogenous replicative stress expected to be present within cells.39 Cells with high levels of endogenous replicative stress may be amenable to sequential administration of PARPi with ATRi or WEE1i, and this approach improved tolerability in relevant ovarian cancer patient-derived xenografts.39
In the clinical setting, a randomized phase II study is currently evaluating adavosertib ± olaparib in patients with recurrent ovarian cancer after progression on a previous PARPi (Table 1), while the OLAPCO study is studying this combination in patients with known P53 or KRAS mutations (Table 1). Yet, overlapping toxicities between adavosertib and PARPi are a concern.11 In a BRCA1/2 wild-type ovarian cancer patient-derived xenograft mouse model, concurrent administration of PARPi and WEE1i led to significant weight loss necessitating termination of therapy after 21 days. In contrast, cycles of sequential PARPi for 1 week followed by WEE1i for 1 week were associated with long-term tolerability and marked prolongation of the depth and duration of tumor shrinkage compared with either inhibitor alone.39 Sequential treatment may retain its clinical efficacy while improving tolerability, therefore the phase I STAR study is evaluating sequential olaparib followed by adavosertib in PARPi-resistant patients with known mutations in DNA damage response genes (Table 1). Limited data are available regarding the clinical efficacy of combinations of PARPi with ATRi or CHK1i. In the ceralasertib plus olaparib arm of study 4, partial response occurred in an ovarian cancer patient who was BRCA-wildtype with preserved ATM expression. Presently, early-phase studies are investigating the use of ATRi and CHK1i in PARPi-resistant patients (Table 1). Therefore, while concurrent and sequential PARPi and ATR–CHK1–WEE1i strategies have entered developmental pipelines, determining optimal combinations and drug schedules in well-selected ovarian cancer subtypes based on their replicative stress profiles will be a crucial factor in advancing these novel combinations into the clinic (Table 1).
Targeting replicative stress in other gynecological cancers
Endometrial cancer
CCNE1 amplification and mutations in PTEN, PIK3CA, ARID1A, ARID5B, and KRAS are frequently found in endometrial cancer and could contribute to increased replicative stress. In endometrial cancer cells, berzosertib increased cell sensitivity to doxorubicin, cisplatin and radiation therapy. The combination of berzosertib with a CHK1i was also noted to be synergistic in-vitro.40 In P53-mutant endometrial cancer cell lines, adavosertib was synergistic with olaparib or gemcitabine and led to increased M-phase cell death indicating mitotic catastrophe.41 This has led to early phase studies of adavosertib in recurrent uterine serous carcinoma (Table 1).
Cervical and vaginal cancer
Cervical and vaginal cancers associated with HPV may harbor virally-mediated defects in CDK function or dysfunctional DNA damage response pathways. Adavosertib has been shown to induce apoptosis in cervical cancer cells displaying high WEE1 expression and to synergize with carboplatin in cervical cancer xenografts.42 Despite encouraging pre-clinical data, a phase I/II study for adavosertib in combination with topotecan or cisplatin in advanced cervical cancer was terminated (NCT01076400).
As cisplatin-based concurrent chemo-radiation therapy represents the backbone of therapy in locally advanced cervical and vaginal cancers, studies are investigating strategies targeting replicative stress in combination with standard chemo-radiotherapy (Table 1). Overactive ribonucleotide reductase is noted to be a driver in the majority of cervical cancers, and triapine was shown to increase replicative stress, arresting cells at G1/S, and resulting in increasing radiation-induced cell kill. A randomized phase II study has reported results of triapine–cisplatin–radiotherapy in locally advanced cervical cancer, and showed trends towards improvements in metabolic complete response (evaluated on positron emission tomography) and 3-year progression-free survival estimates for patients receiving triapine–cisplatin–radiation compared with cisplatin–radiation alone. No significant differences in the rate of adverse events were reported.43 This has led to a phase III study in locally advanced cervical and vaginal cancer (Table 1).
Challenges and future directions
Early efforts to target replicative stress in gynecological cancers have elucidated important challenges and highlighted the potential for novel combinations with other drugs. Undoubtedly, a major challenge will be determining the optimal dose and schedule of these agents that balances tolerability with efficacy. The key dose-limiting toxicity of ATRi–CHK1i–WEE1i seen in early phase clinical trials has been dose-dependent myelosuppression (Table 1), and this has made combining these drugs with cytotoxic chemotherapies particularly challenging, sometimes compromising treatment efficacy. For example, the recommended phase II dose of the ATRi M6620 when combined with carboplatin is a quarter of the dose intensity compared with the recommended M6620 monotherapy dose.44 45 Other significant high grade toxicities that have been reported include drug-induced liver dysfunction for ceralasertib46 and SRA737,47 as well as high grade pulmonary toxicity with triapine use.48 Although dose-limiting cardiac toxicities were reported in 4.7% of patients enrolled on a phase I dose-escalation study of the CHK1/2 inhibitor AZD7762 (AstraZeneca) in combination with gemcitabine,49 praxasertib has not been associated with this feared side effect, even at the maximal tolerated dose.50 As we have learned from difficulties combining high-potency PARPi with chemotherapy due to toxic myelosuppression, the sequential administration of chemotherapy followed by maintenance PARPi has led to clinical approvals, and this may be the required approach for novel ATRi–WEE1i–CHK1i.
Another area that deserves further exploration is the crosstalk between enhanced replicative stress with the immune response. Replicative stress-related DNA damage may lead to increased cytosolic DNA fragments, thus triggering the cyclic guanosine monophosphate–AMP synthase–stimulator of interferon genes (cGAS-STING) pathway, which drives an innate immune response. In immunocompetent advanced prostate cancer models, inhibition of ATR led to increased S-phase DNA damage and cGAS-STING activation together with increased levels of type I interferon, C-X-C motif chemokine ligand 10 (CXCL10), and C-C motif chemokine ligand 5 (CCL5), which are transcriptional targets of interferon regulatory factor 3 (IRF3), indicating an innate immune response. Similar results have been reported in small cell lung cancer models treated with the CHK1 inhibitor SRA737,51 indicating the potential immunomodulatory role of ATR/CHK1i. The combination of anti-programmed death-ligand 1 (PD-L1) therapy with SRA737 was also shown to be synergistic compared with either agent alone, resulting in complete tumor growth inhibition in small cell lung cancer mouse models.51 In breast cancer samples, a DNA damage response-deficient molecular subtype was associated with increased CD4+ and CD8+ immune cell infiltration and constitutive cGAS-STING activation.52 S-phase DNA damage led to increased PD-L1 expression and was STING-dependent.52 In this vein, several studies are investigating the combination of programmed cell death protein 1 (PD-1)/PD-L1 immune checkpoint inhibitors in combination with inhibitors of DNA-PKcs, CDK4/6, ATR, and WEE1. This strategy also has the added advantage of non-overlapping toxicity profiles between drug classes of immune checkpoint inhibitors and cell cycle checkpoint blockade.53 Of note, a phase Ib/II study of avelumab, carboplatin, and berzosertib in PARPi-resistant ovarian cancer has recently completed and results are awaited (Table 2).
Selected clinical trials of cell cycle checkpoint inhibitors with PD-1/PD-L1 immune checkpoint blockade relevant to gynecological cancers
The identification of elevated reactive oxygen species as a mechanism for inducing endogenous replicative stress also indicates the interplay between replicative stress and metabolic pathways. Intracellular reactive oxygen species may be increased by reducing glutathione synthesis, inhibiting the cysteine transporter, or inactivation of nuclear factor erythroid 2-related factor 2 (NRF2)-dependent reductive pathways. These may be potential targets to enhance intracellular replicative stress. Furthermore, glycolysis supplies intermediates for biosynthesis of nucleotides via the pentose-phosphate pathway,54 and the inhibition of glycolytic intermediates such as through 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) inhibitors or others may logically affect ribonucleotide supply in the setting of high demand at stalled forks and increase replicative stress. These entry points could be interesting areas for co-targeting of metabolic pathways to enhance replicative stress in cancer therapy.
Glaringly, at present, no confirmed biomarkers for strategies targeting replicative stress in gynecological cancers have been clearly identified. However, early phase studies have called attention to several emerging predictive markers for patient selection (Table 3). P53 mutations are hypothesized to lead to increased reliance on the G2/M checkpoint, therefore increasing sensitivity to ATRi–WEE1i–CHK1i. Yet, in-vitro data have been conflicting with respect to the predictive ability of P53 mutations for ATRi sensitivity in various cell lines, likely relating to the wide range of mutations and their diverse functional consequences in different cellular contexts.55 ATM loss has been associated with sensitivity to ATRi and is hypothesized to increase tumor dependency on ATR. Anecdotal reports of response in patients with ATM loss have been described in early phase studies, and some trials have incorporated ATM loss into patient selection criteria.11 Yet, the optimal method for determining ATM loss or other genomic markers remains contentious. A phase III study of paclitaxel ± olaparib in patients with advanced gastric cancer who were ATM-negative based on immunohistochemistry (using the Ventana ATM Y170 assay) failed to show improvement in clinical outcomes for patients receiving olaparib compared with placebo,56 garnering questions regarding the optimal choice of assay and threshold cut-off points that should be used in patient selection. Next-generation tumor sequencing offers the opportunity to screen patients to identify DNA damage response mutations, homologous recombination deficiency, and P53 mutations, among others, but the functional significance of individual mutations may be challenging to interpret. Multiple genomic aberrations may be required to produce the high replicative stress phenotype for exploitation, and their interaction may not be easily discerned from tumor profiling alone. Clinical trials in these subgroups of patients are ongoing (Table 1), and will be required to inform the usefulness of these markers in gynecological cancers. Further experimental assays that are being utilized pre-clinically to detect replicative stress in tumors are shown in Table 4.
Predictive genomic and epigenetic biomarkers of replicative stress in pre-clinical development
Experimental assays that are used for the detection of replicative stress
In conclusion, targeting replicative stress represents a fast emerging area of therapy in gynecological cancers, and multiple possibilities remain on the horizon with respect to novel means of enhancing through rational treatment combinations. The success of efforts to identify predictive biomarker assays, understand resistance mechanisms, and optimize clinically feasible drug schedules will be crucial to enable the addition of replicative stress targeting to our treatment armamentarium.
References
Footnotes
Contributors Conceptualization: NYLN, DT. Data curation: NYLN. Original draft preparation: NYLN, VS. Review and editing: NYLN, VS, DT. Visualization: NYLN, VS, DT. Supervision: DT. Project administration: NYLN.
Funding DT is supported by the National Medical Research Council, Singapore (CSAINV16may008), NYLN is supported by the National Medical Research Council, Singapore (MOH-FLWSHP19may-0006).
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.