Article Text

Abstract
Objective To report results from an integrated efficacy and safety analysis supporting the European Commission's approval of the poly(ADP-ribose) polymerase inhibitor rucaparib as monotherapy treatment for relapsed, platinum-sensitive, BRCA-mutated ovarian cancer.
Methods Efficacy was analyzed in platinum-sensitive patients from Study 10 (NCT01482715) and ARIEL2 (NCT01891344) who had high-grade serous or endometrioid epithelial ovarian, fallopian tube, or primary peritoneal cancer and a deleterious BRCA1 or BRCA2 mutation and received two or more prior chemotherapies (including two or more platinum-based therapies). The primary end point was investigator-assessed, confirmed objective response rate (visit cut-off: April 10, 2017). Safety was analyzed in patients with ovarian cancer, regardless of BRCA mutation status or lines of prior chemotherapies, who received at least one dose of rucaparib 600 mg in either study (visit cut-off: December 31, 2017).
Results In the integrated platinum-sensitive efficacy population (n=79), objective response rate was 64.6% (95% CI, 53.0 to 75.0); 10.1% (8/79) of patients had a complete response and 54.4% (43/79) had a partial response. Median duration of response was 294 days (95% CI, 224 to 393). In the integrated safety population (n=565), the most common any-grade treatment-emergent adverse events were nausea (77.7%, 439/565), asthenia/fatigue (74.7%, 422/565), vomiting (45.8%, 259/565), and hemoglobin decreased (44.2%, 250/565). Treatment-emergent adverse events led to treatment interruption, dose reduction, or discontinuation in 60.2% (340/565), 46.0% (260/565), and 16.8% (95/565) of patients.
Conclusions In patients with platinum-sensitive, BRCA-mutated ovarian cancer, rucaparib demonstrated antitumor activity and is the first and currently the only poly(ADP-ribose) polymerase inhibitor approved by the European Commission as treatment for this population. The safety analysis used a more recent visit cut-off date and larger population than previously published, was consistent with prior reports, and was the basis for the treatment-indication safety population in rucaparib’s recently updated European Union label.
- ovarian cancer
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
HIGHLIGHTS
Patients (65%) with platinum-sensitive, BRCA-mutated ovarian cancer responded to rucaparib treatment.
The integrated efficacy data supported the approval of rucaparib treatment in the European Union.
In the updated safety analysis, rucaparib had a manageable safety profile similar to prior reports.
Introduction
In Europe, ovarian cancer accounted for 3.4% of new cancer cases and 5.2% of cancer deaths in women in 2018.1 Although most patients respond to initial treatment (surgery followed by platinum-based chemotherapy with or without bevacizumab) the majority will relapse.2 Most patients with recurrent ovarian cancer will receive additional lines of platinum-based chemotherapy; however, this may be limited by cumulative chemotherapy-related toxicities. In particular, 44% of patients receiving third-line platinum-based chemotherapy develop platinum hypersensitivity, with patients having a BRCA mutation being at higher risk.3 4
Rucaparib (formerly known as CO-338, AG-014447, and PF-01367338) is an oral, small molecule inhibitor of poly(ADP-ribose) polymerase 1, 2, and 3.5 These enzymes are crucial for the repair of single-strand breaks in DNA. Rucaparib blocks normal DNA repair mechanisms by binding to the poly(ADP-ribose) polymerase catalytic domain5 and may also cause poly(ADP-ribose) polymerase enzymes to remain bound to damaged DNA.6 Cells with defects in homologous recombination, a process through which double-strand breaks in DNA are repaired, are particularly sensitive to poly(ADP-ribose) polymerase inhibition due to synthetic lethality,7 8 and rucaparib has demonstrated antitumor activity in tumors with homologous recombination deficiency.9–12 Up to half of high-grade serous ovarian cancers (including fallopian tube and primary peritoneal cancers) may exhibit homologous recombination deficiency at diagnosis, with 18% harboring a germline BRCA1 or BRCA2 (BRCA1/2) mutation, 7% a somatic BRCA1/2 mutation, and 20% a mutation in, or epigenetic silencing of, another homologous recombination gene.13 14
Rucaparib has been approved by the European Commission for use as monotherapy in patients with platinum-sensitive, relapsed, or progressive, BRCA-mutated (germline and/or somatic), high-grade epithelial ovarian, fallopian tube, or primary peritoneal cancer who have been treated with two or more prior lines of platinum-based chemotherapy and are unable to tolerate further platinum-based chemotherapy.15 Currently, it is the only poly(ADP-ribose) polymerase inhibitor approved in the European Union for use in patients with ovarian cancer in the treatment setting. Rucaparib was also recently approved by the European Commission as monotherapy for the maintenance treatment of patients with platinum-sensitive, relapsed, high-grade epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in response (complete or partial) to platinum-based chemotherapy.15
Here, we report an integrated efficacy analysis based on two studies, Study 10 (CO-338-010; NCT01482715)10 and ARIEL2 (CO-338-017; NCT01891344),11 that demonstrated the antitumor activity of rucaparib as treatment in patients with platinum-sensitive, recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer with a deleterious BRCA1/2 mutation and supported the recent European Commission approval of rucaparib in the treatment setting. We also provide an updated integrated safety analysis based on these two studies that utilizes a later visit cut-off (December 31, 2017) and larger population of patients with ovarian cancer (n=565) than previously reported (April 29, 2016; n=377).16 This analysis served as the basis for the treatment indication safety population described in the recently updated label for rucaparib in the European Union.15
Methods
Constituent study designs
Data in this manuscript are derived from patients enrolled in Study 10 or ARIEL2. Study 10 is a three-part, open-label, phase 1/2 study of oral rucaparib given until disease progression or unacceptable toxicity.10 ARIEL2 is a two-part, phase 2, open-label study of oral rucaparib 600 mg twice daily given until disease progression, unacceptable toxicity, or death in patients with relapsed high-grade ovarian cancer.11 Key study design and patient eligibility details are provided in the online Supplementary Methods (Supplemental Digital Content 1); full methodologies for each study have been published elsewhere.10 11 Each study was approved by the independent review board at each participating site and carried out in accordance with the Declaration of Helsinki and the International Conference on Harmonization’s Good Clinical Practice Guidelines. Patients provided written informed consent before participation.
Supplemental material
Integrated analysis datasets
For the integrated efficacy analysis, the study enrollment cut-off was October 1, 2015, and the visit cut-off was April 10, 2017. For the updated integrated safety analysis, the visit cut-off was December 31, 2017; at that time, both studies were fully enrolled (last patient enrolled August 31, 2016).
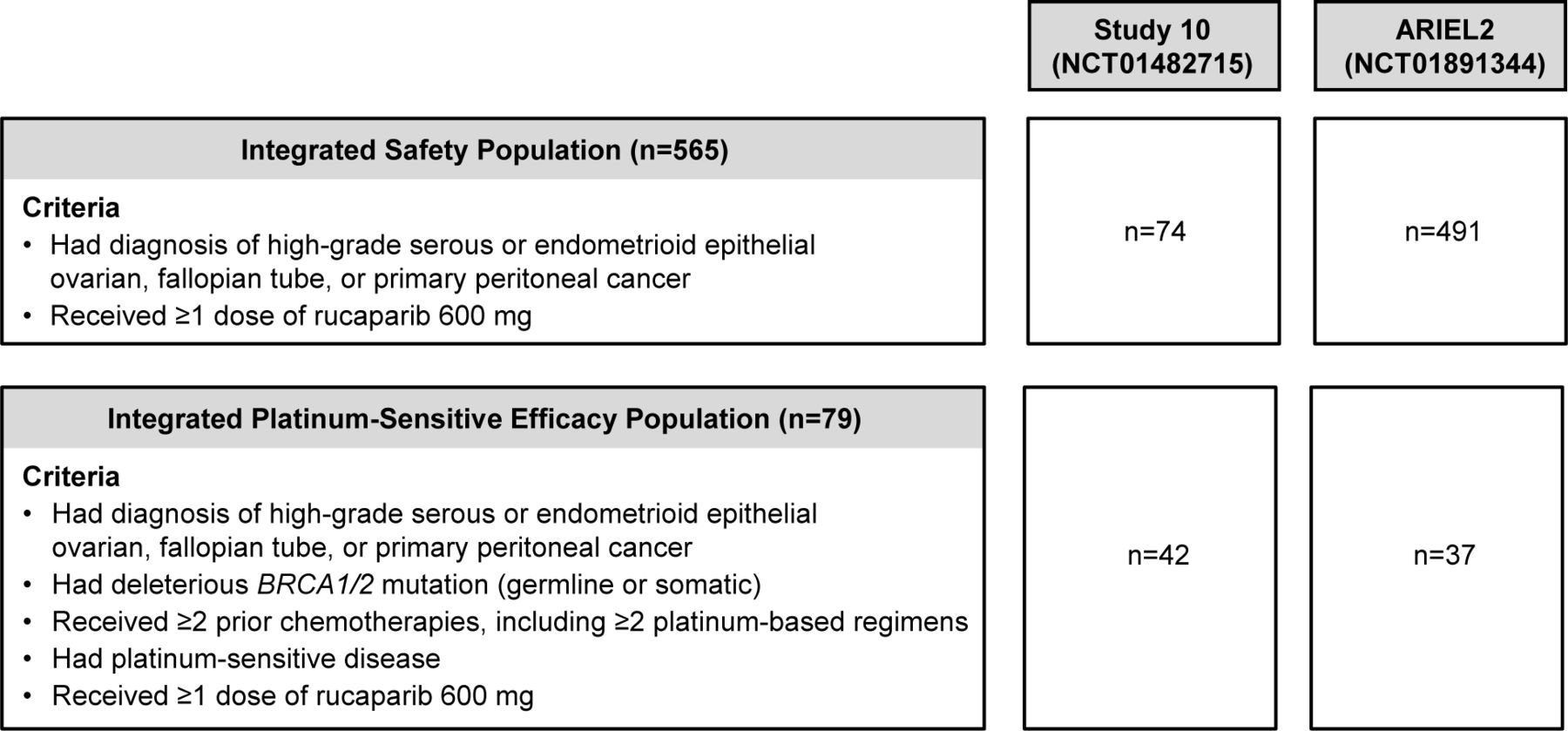
The integrated platinum-sensitive efficacy population included patients from Study 10 (Part 2A only) and ARIEL2 (Parts 1 and 2) who met the following eligibility criteria: a diagnosis of high-grade serous or endometrioid epithelial ovarian, fallopian tube, or primary peritoneal cancer; a deleterious BRCA1/2 mutation (germline BRCA1/2 mutation in Study 10, germline or somatic BRCA1/2 mutation in ARIEL2); at least two prior chemotherapies, including at least two platinum-based therapies; platinum-sensitive disease (disease progression ≥6 months after last platinum); and at least one dose of rucaparib 600 mg (Figure 1).

Integrated safety population and integrated platinum-sensitive efficacy population. The visit cut-off for the integrated safety analysis was December 31, 2017, and the visit cut-off for the integrated platinum-sensitive efficacy analysis was April 10, 2017.
The integrated safety population included all patients with ovarian cancer from Study 10 (Parts 1, 2A, 2B, and 3) and ARIEL2 (Parts 1 and 2) who took at least one dose of rucaparib 600 mg. Patients were included in the integrated safety population irrespective of BRCA1/2 mutation status, number or type of prior therapies, and prior response to platinum therapy.
Integrated platinum-sensitive efficacy analysis outcomes
The primary outcome of interest in the integrated platinum-sensitive efficacy analysis was investigator-assessed confirmed objective response rate per Response Evaluation Criteria In Solid Tumors version 1.1 (RECIST), defined as the proportion of patients with a confirmed complete response or partial response on subsequent tumor assessment ≥28 days after the first response documentation.17 The objective response rate is presented with 95% CIs calculated using Clopper–Pearson methodology. Secondary end points included investigator-assessed best response in the sum of target lesions, duration of response, and progression-free survival. Secondary end point definitions are provided in the online Supplementary Methods (Supplemental Digital Content 1). All analyses are presented descriptively.
Integrated safety analysis outcomes
In both trials, safety assessments included adverse event monitoring and laboratory investigations. Verbatim terms were coded using the Medical Dictionary for Regulatory Activities version 19.1.18 Adverse event severities and laboratory abnormalities were classified according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03.19 Safety outcomes of interest were treatment-emergent adverse events of all grades, grade 3 or greater treatment-emergent adverse events, serious adverse events, treatment-related adverse events, and adverse events leading to dose modification (treatment interruption and/or dose reduction), treatment discontinuation, and death. A treatment-emergent adverse event was defined as an adverse event with an onset date on or after the date of first dose of study medication until the date of the last study medication dose plus 28 days.
Results
Integrated platinum-sensitive efficacy population
The integrated platinum-sensitive efficacy population included 79 patients from Study 10 Part 2A (n=42) and ARIEL2 Parts 1 (n=24) and 2 (n=13) (Figure 1). Median age was 59 years (range, 33–84), and the majority of patients had epithelial ovarian cancer (87.3%, 69/79) (Table 1). All patients had a deleterious germline (83.5%, 66/79) or somatic (16.5%, 13/79) BRCA1/2 mutation.
Baseline patient characteristics and prior chemotherapy in the integrated, platinum-sensitive efficacy population and the integrated safety population
Integrated platinum-sensitive efficacy findings
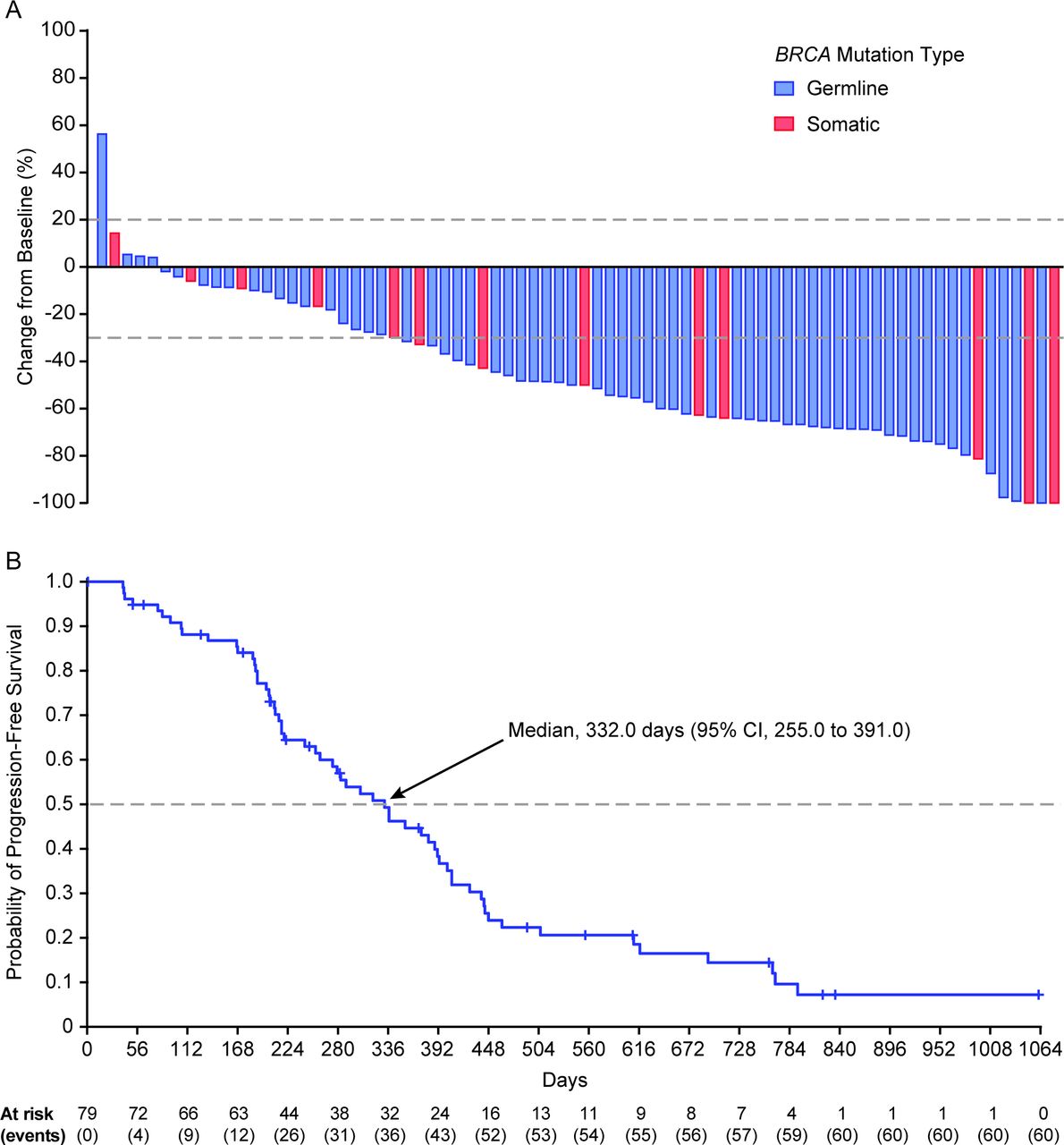
In the integrated platinum-sensitive efficacy population, the investigator-assessed confirmed objective response rate by RECIST was 64.6% (95% CI, 53.0 to 75.0) (Table 2). Best overall investigator-assessed confirmed complete and partial responses were reported for 10.1% (8/79) and 54.4% (43/79) of patients, respectively; 25.3% (20/79) of patients had stable disease and 5.1% (4/79) had progressive disease. Efficacy results were similar among the subgroups of patients with a germline BRCA1/2 mutation and those with a somatic BRCA1/2 mutation, with investigator-assessed objective response rates of 65.2% (95% CI, 52.4 to 76.5) and 61.5% (95% CI, 31.6 to 86.1), respectively. Most patients in the overall population (89.9%, 71/79) had a decrease from baseline in the sum of the diameter of target lesions, with most decreases being ≥30% (Figure 2A), further supporting the antitumor activity of rucaparib. In the integrated platinum-sensitive efficacy population, the median duration of investigator-assessed confirmed response was 294 days (95% CI, 224 to 393) (Table 2). Median investigator-assessed progression-free survival was 332 days (95% CI, 255 to 391) (Table 2; Figure 2B); 24.1% (19/79) of patients had not progressed at the time of the visit cut-off. At the time of this analysis, overall survival data were not mature (censoring rate, 86.1% (68/79)).

{kind=link}
{kind=link}
A: Best percentage change from baseline for sum of diameter of target lesions in the integrated platinum-sensitive efficacy population according to whether patients had a germline or somatic BRCA mutation. Each bar represents a single patient; the upper dotted line indicates the threshold for progressive disease, a 20% increase in the sum of the longest diameter of the target lesions, whereas the lower dotted line indicates the threshold for partial response, a 30% decrease in the sum of the longest diameter of the target lesions. B: Investigator-assessed progression-free survival in the integrated platinum-sensitive efficacy population. Progression-free survival was analyzed by Kaplan–Meier methodology; data were censored at the last tumor assessment for patients without documented progression.
Investigator-assessed confirmed objective response rate (per RECIST), duration of response, and progression-free survival in the platinum-sensitive efficacy population
Integrated safety population
The integrated safety population included 565 patients with epithelial ovarian, fallopian tube, or primary peritoneal cancer from Study 10 Parts 1, 2A, 2B, and 3 (n=74) and ARIEL2 Parts 1 and 2 (n=491) (Figure 1). The median duration of rucaparib treatment in the integrated safety population was 5.3 months (range, 0.1–44.5).
Baseline median age was 63 years (range, 31–91) (Table 1). More than one-third of patients (37.2%, 210/565) had a deleterious BRCA1/2 mutation (germline 27.4%, 155/565; somatic 8.1%, 46/565).
Integrated safety findings
All patients in the integrated safety population had at least one treatment-emergent adverse event, and 63.2% (357/565) had a grade 3 or greater event. The most frequently reported treatment-emergent adverse events were nausea (77.7%, 439/565), asthenia/fatigue (74.7%, 422/565), vomiting (45.8%, 259/565), hemoglobin decreased (44.2%, 250/565), and alanine/aspartate aminotransferase increased (39.5%, 223/565) (Table 3). Elevations in alanine/aspartate aminotransferase occurred within the first few weeks of rucaparib treatment, were reversible, and were rarely associated with increases in bilirubin. Frequent grade 3 or greater treatment-emergent adverse events included hemoglobin decreased (24.2%, 137/565), asthenia/fatigue (11.3%, 64/565), and alanine/aspartate aminotransferase increased (10.8%, 61/565). Myelodysplastic syndrome/acute myeloid leukemia was reported as a treatment-emergent adverse event in 0.4% (2/565) of patients as of the December 31, 2017, visit cut-off.
Safety summary: all patients from Study 10 or ARIEL2 who received at least one dose of rucaparib 600 mg twice daily
Treatment-emergent adverse events led to treatment interruption in 60.2% (340/565) of patients and dose reduction in 46.0% (260/565) (Table 3). Those most frequently leading to dose modification (treatment interruption and/or dose reduction) included hemoglobin decreased (22.1%, 125/565), asthenia/fatigue (21.4%, 121/565), nausea (17.0%, 96/565), platelets decreased (13.1%, 74/565), vomiting (12.7%, 72/565), and alanine/aspartate aminotransferase increased (8.0%, 45/565). Excluding disease progression, treatment-emergent adverse events led to treatment discontinuation in 16.8% (95/565) of patients (Table 3). Of these, the most frequent were asthenia/fatigue (3.0%, 17/565), small intestinal obstruction (1.9%, 11/565), platelets decreased (1.6%, 9/565), hemoglobin decreased (1.6%, 9/565), nausea (1.2%, 7/565), vomiting (1.2%, 7/565), abdominal pain (0.9%, 5/565), and ascites (0.9%, 5/565).
A treatment-emergent adverse event with an outcome of death occurred in 4.6% (26/565) of patients. Of these deaths, 2.8% (16/565) were associated with malignant neoplasm progression. Ten patient deaths (1.8%) were associated with nonprogression events. This included one resulting from B-cell lymphocytic leukemia, which was assessed by the investigator as related to rucaparib, and nine resulting from events assessed by the investigator as unrelated to rucaparib, with the primary reasons reported as general physical health deterioration (0.7%, 4/565), cerebral artery embolism (0.2%, 1/565), cerebrovascular accident (0.2%, 1/565), intestinal obstruction (0.2%, 1/565), sepsis (0.2%, 1/565), and septic shock (0.2%, 1/565).
Discussion
In the integrated platinum-sensitive efficacy population, almost two-thirds of patients with platinum-sensitive ovarian cancer had a confirmed objective response to treatment with rucaparib. These data reflect confirmed responses in representative target lesions and all other nontarget lesions (if present) and also account for disease progression (eg, growth of target/nontarget lesions or the appearance of a new lesion). It is complemented by the examination of best response in the sum of target lesions, which demonstrated the maximal depth of response achieved for each patient, with a majority of patients having a decrease in target lesion size from baseline. Furthermore, the response was durable (median duration of response, 9.7 months (95% CI, 7.4 to 12.9)). Together, the integrated platinum-sensitive efficacy data support the antitumor activity of rucaparib in this patient population. In 2017, Oza et al reported an integrated efficacy analysis supporting the initial approval of rucaparib in the United States for the treatment of adult patients with deleterious BRCA mutation (germline and/or somatic)-associated epithelial ovarian, fallopian tube, or primary peritoneal cancer who have been treated with two or more chemotherapies.16 The Oza et al study integrated efficacy population consisted of 106 patients with ovarian cancer from Study 10 and ARIEL2, which included seven patients with platinum-refractory ovarian cancer and 20 with platinum-resistant ovarian cancer who were not included in our current efficacy analysis.16 In an updated analysis, the resulting objective response rate was 54.7% (58/106) in the overall population and 35.0% (7/20) in patients with platinum-resistant ovarian cancer, and there were no responses among patients with platinum-refractory ovarian cancer.15
The current integrated safety analysis demonstrated that rucaparib has a manageable safety profile consistent with those of other poly(ADP-ribose) polymerase inhibitors.20–30 Gastrointestinal events, hematological toxicities, and fatigue were among the more frequently observed treatment-emergent adverse events. The treatment-emergent adverse events and laboratory abnormalities were managed with treatment interruption, treatment modification, and/or supportive care. This integrated safety analysis included 188 more patients with ovarian cancer, with an additional 20 months of follow-up than previously reported in Oza et al (n=377; visit cut-off: April 29, 2016),16 and no new safety signals were identified. Notably, the incidence of myelodysplastic syndrome/acute myeloid leukemia in this integrated safety analysis population was consistent with the 0.5% reported in a separate, larger analysis that included 1321 patients, regardless of tumor type, who received at least one dose of oral rucaparib in a clinical study in either the treatment or maintenance setting.15
Rucaparib is currently the only poly(ADP-ribose) polymerase inhibitor approved in the treatment setting in the European Union;15 21 22 rucaparib was also recently approved by the European Commission for use in the maintenance setting.15 Rucaparib’s approval in the treatment setting provides an alternative to third-line or later chemotherapy. Studies evaluating the use of platinum and nonplatinum chemotherapies beyond second-line treatment are limited. In single-center, retrospective studies, objective responses to third-line chemotherapy ranged from 12% to 41%.31–33 The majority of patients in these studies received carboplatin, paclitaxel, or topotecan. Although these studies did not report on the incidence of adverse events, the safety profiles of these drugs are well established, with the most common adverse events being hematological toxicities (eg, neutropenia and anemia), neurotoxicity, and alopecia.34 Furthermore, there is a need for alternatives to chemotherapy in later settings as additional platinum-based chemotherapy may be unsuitable for certain patients due to cumulative chemotherapy-related toxicities and platinum hypersensitivity. Platinum hypersensitivity develops in approximately 8% to 44% of patients,4 35–37 with the risk increasing with the number of prior cycles of platinum-based chemotherapy.4 35 37
The poly(ADP-ribose) polymerase inhibitors niraparib and olaparib are only approved in the maintenance setting in the European Union for use in patients with high-grade serous epithelial ovarian, fallopian tube, or primary peritoneal cancer and a platinum-sensitive relapse who are in response (complete or partial) to platinum-based chemotherapy.21 22 In the United States, olaparib is approved as fourth-line or later treatment of patients with germline only BRCA-mutated, advanced ovarian cancer based on data from the phase 2 trial, Study 42 (NCT01078662).23 27 In Study 42, the objective response rate in patients who had received three or more prior lines of chemotherapy was 34% (46/137), and median duration of response was 7.9 months. Of the 46 patients with a confirmed response, 52% (24/46) were platinum resistant and 39% (18/46) were platinum sensitive. The most common adverse events in Study 42 were nausea (60%), fatigue (55%), vomiting (44%), and anemia (34%).27 Niraparib has also been examined in the treatment setting26 38 but is not yet approved for this indication. In the phase 2, open-label QUADRA study (NCT02354586), 27% (14/51) of patients with platinum-sensitive disease who had received three or more regimens and were homologous recombination deficient had an objective response to niraparib; the median duration of response was 9.2 months.38 In QUADRA, the most common grade 3 or greater adverse events across all patients were thrombocytopenia (28%), anemia (25%), and neutropenia (12%).38
A limitation of the current analysis is that data are only from open-label, single-arm, nonrandomized, phase 2 trials. Randomized data are not yet available for rucaparib in the treatment setting. However, a phase 3 trial is underway to further evaluate rucaparib versus standard-of-care chemotherapy in patients with relapsed, platinum-sensitive or platinum-resistant ovarian cancer with a BRCA1/2 mutation in the treatment setting (ARIEL4; NCT02855944). In a report of a randomized, phase 3 trial in patients with relapsed, BRCA-mutated, platinum-sensitive ovarian cancer (SOLO3; NCT02282020), olaparib demonstrated significant improvement over single-agent, nonplatinum chemotherapy in objective response rate (primary end point; 72% vs 51%) and progression-free survival (secondary end point; median, 13.4 vs 9.2 months (HR, 0.62; 95% CI, 0.43 to 0.91)) in the treatment setting; safety and tolerability were consistent with those in prior studies.39
In summary, the results reported here demonstrate that rucaparib has robust antitumor activity in patients with platinum-sensitive ovarian cancer associated with a BRCA mutation. On the basis of these results, the European Commission has granted approval for rucaparib as monotherapy treatment of adult patients with platinum-sensitive, relapsed or progressive, BRCA-mutated (germline and/or somatic), high-grade epithelial ovarian, fallopian tube, or primary peritoneal cancer who have been treated with two or more prior lines of platinum-based chemotherapy and are unable to tolerate further platinum-based chemotherapy. Furthermore, the results from the updated safety analysis in patients with ovarian cancer demonstrate that rucaparib has a tolerable and manageable safety profile that is consistent with those of prior reports of rucaparib and other poly(ADP-ribose) polymerase inhibitors.
Acknowledgments
We thank all of the patients who participated in ARIEL2 or Study 10, as well as their families and caregivers. We also thank the investigators for their contributions to the administration and execution of the studies, Foundation Medicine, Inc., for their contributions, and the Clovis Oncology study teams for clinical development and operational support.
References
Footnotes
Contributors RSK, RS-F, IAM, and LM conceived and designed the studies. RSK, AO, IR-C, AL, JB, YD, AMO, RS-F, SMD, TC, LM, SG, DL, JAL, and IAM were involved in acquisition, analysis, or interpretation of data for the work. RSK, AO, IR-C, AL, JB, YD, AMO, RS-F, SMD, TC, LM, SG, DL, JAL, and IAM were involved in drafting the manuscript and provided critical feedback. RSK, AO, IR-C, AL, JB, YD, AMO, RS-F, SMD, TC, LM, SG, DL, JAL, and IAM approved the final manuscript.
Funding This work was funded by Clovis Oncology, Inc. Authors who are employees of Clovis Oncology were involved in the design of the studies reported in this publication, as well as the analysis and interpretation of the data, writing of the report, and approval of the paper for publication. The sponsor was responsible for the collection, analysis, and interpretation of the data. One or more of the trials discussed in this publication were supported by the NIHR/Wellcome UCH Clinical Research Facility and Experimental Cancer Medicine Centres at UCL, Newcastle, and Imperial. RSK is supported in part by the UCH/UCL Biomedical Research Centre. JAL is an NIHR Senior Investigator. Writing and editorial assistance funded by Clovis Oncology was provided by Nathan Yardley, PhD, Mollie Marko, PhD, and Shannon Davis of Ashfield Healthcare Communications (Middletown, CT).
Competing interests RSK has served on advisory boards for Clovis Oncology, Roche, and Tesaro. AO has served on advisory boards for Clovis Oncology, AstraZeneca, Genmab/Seattle Genetics, ImmunoGen, PharmaMar, Roche, and Tesaro; has received support for travel or accommodation from Clovis Oncology, AstraZeneca, PharmaMar, and Roche; and reports institutional research grant support from Clovis Oncology, AbbVie Deutschland, Ability Pharmaceuticals, Advaxis, Aeterna Zentaris, Amgen, Aprea Therapeutics, Eisai, ImmunoGen, Merck/Merck Sharp & Dohme, Millennium Pharmaceuticals, PharmaMar, Roche, and Tesaro. IR-C has served on advisory boards for Clovis Oncology, AstraZeneca, Genmab/Seattle Genetics, ImmunoGen, PharmaMar, Roche, and Tesaro and received support for travel or accommodation from AstraZeneca and Roche. AL has served on advisory boards for Clovis Oncology, AstraZeneca, Gritstone, Genmab/Seattle Genetics, and Tesaro; reports institutional support for clinical trials from Clovis Oncology and AstraZeneca; and reports boarding and travel expenses for congress activities from Clovis Oncology, AstraZeneca, and Roche. JB has served on advisory boards for Clovis Oncology, AstraZeneca, and Bristol-Myers Squibb and received support for travel from AstraZeneca. YD has served on advisory boards for Clovis Oncology, AstraZeneca, Genmab, and Tesaro; serves on a trial steering committee for AstraZeneca; has received research funding from Clovis Oncology; her institution, Newcastle Hospitals NHS Foundation Trust, has received reimbursement of study costs from Clovis Oncology for a clinical trial described in this manuscript. YD and her employer (Newcastle University) receive royalties in recognition of their role in the preclinical development of rucaparib. AMO has served on steering committees for Clovis Oncology, AstraZeneca, and Tesaro. RS-F has served on advisory boards for Clovis Oncology. SMD has received honoraria from Clovis Oncology, AstraZeneca, and Bristol-Myers Squibb, and her institution has received research funding from Clovis Oncology and AstraZeneca. TC, LM, and SG are employees of Clovis Oncology and may own stock or have stock options in that company. DL has served in a consulting or advisory role for Clovis Oncology, AstraZeneca, Genmab, Merck, and Tesaro and received institutional research support from Clovis Oncology, Merck, PharmaMar, and Tesaro. JAL has served in an advisory role for Clovis Oncology, Artios Pharma, AstraZeneca, Merck/Merck Sharp & Dohme, Pfizer, Seattle Genetics, and Tesaro; is chair of an independent Data Monitoring Committee for Regeneron; and has served on speakers' bureaus for and received research grants from AstraZeneca and Merck/Merck Sharp & Dohme. IAM has served on advisory boards for Clovis Oncology, AstraZeneca, Takeda, and Tesaro and receives institutional funding from AstraZeneca.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement For more information about this manuscript, please contact the corresponding author. Data that are summarized in this manuscript are from the clinical studies ARIEL2 (NCT01891344) and Study 10 (NCT01482715), both of which were sponsored by Clovis Oncology, Inc. (Boulder, CO). Efficacy data in this manuscript are summarized for patients who enrolled on ARIEL2 or Study 10 by October 1, 2015. Safety follow-up data are included for the period ending on December 31, 2017. For more detailed information about these studies or the data summarized herein, please contact: medinfo@clovisoncology.com.